MED23遺伝子の変異の同定および転写の異常と知的障害とのかかわり
橋本 悟
(フランスInstitute of Genetics and Molecular and Cellular Biology,Program of Functional Genomics & Cancer)
email:橋本 悟
DOI: 10.7875/first.author.2011.135
MED23 mutation links intellectual disability to dysregulation of immediate early gene expression.
Satoru Hashimoto, Sarah Boissel, Mohammed Zarhrate, Marlène Rio, Arnold Munnich, Jean-Marc Egly, Laurence Colleaux
Science, 333, 1161-1163 (2011)
MED23は遺伝子発現において重要な役割を担うメディエーター複合体のサブユニットのひとつである.筆者らは,常染色体劣勢の遺伝様式を示す知的障害者の家系より,新規にMED23遺伝子の変異を同定した.この変異では転写因子からの情報をRNAポリメラーゼIIに伝達するというメディエーター複合体の機能が阻害されており,血清による刺激ののちのJUN遺伝子およびFOS遺伝子を含む一部の即時発現型遺伝子の発現調節に異常を認めた.さらに,これらの即時発現型遺伝子における発現調節の異常は,知的障害をきたすことの知られる,ほかのメディエーター複合体のサブユニットや基本転写因子の変異においても確認された.これらのことは,中枢神経系の発達においてメディエーター複合体が重要であることを示すとともに,即時発現型遺伝子における発現調節の異常が認知障害の指標となりうることを示唆した.
真核生物においてmRNAはRNAポリメラーゼIIのみにより合成(転写)されるが,その過程は多くの転写因子により制御されている.細胞の内外に生じるさまざまな刺激は転写因子を活性化させ,転写因子は遺伝子の上流のプロモーター領域にある特異的な配列に結合する.結合した転写因子はその下流に位置するRNAポリメラーゼIIおよび付属する基本転写因子に刺激を伝達するが,この刺激の伝達により転写の開始することが知られている.メディエーター複合体は転写因子とRNAポリメラーゼIIとのあいだの架け橋としてはたらき,多くの転写因子と結合することにより巨大なタンパク質ネットワークを形成することで複雑な転写調節が行われている1)(図1).
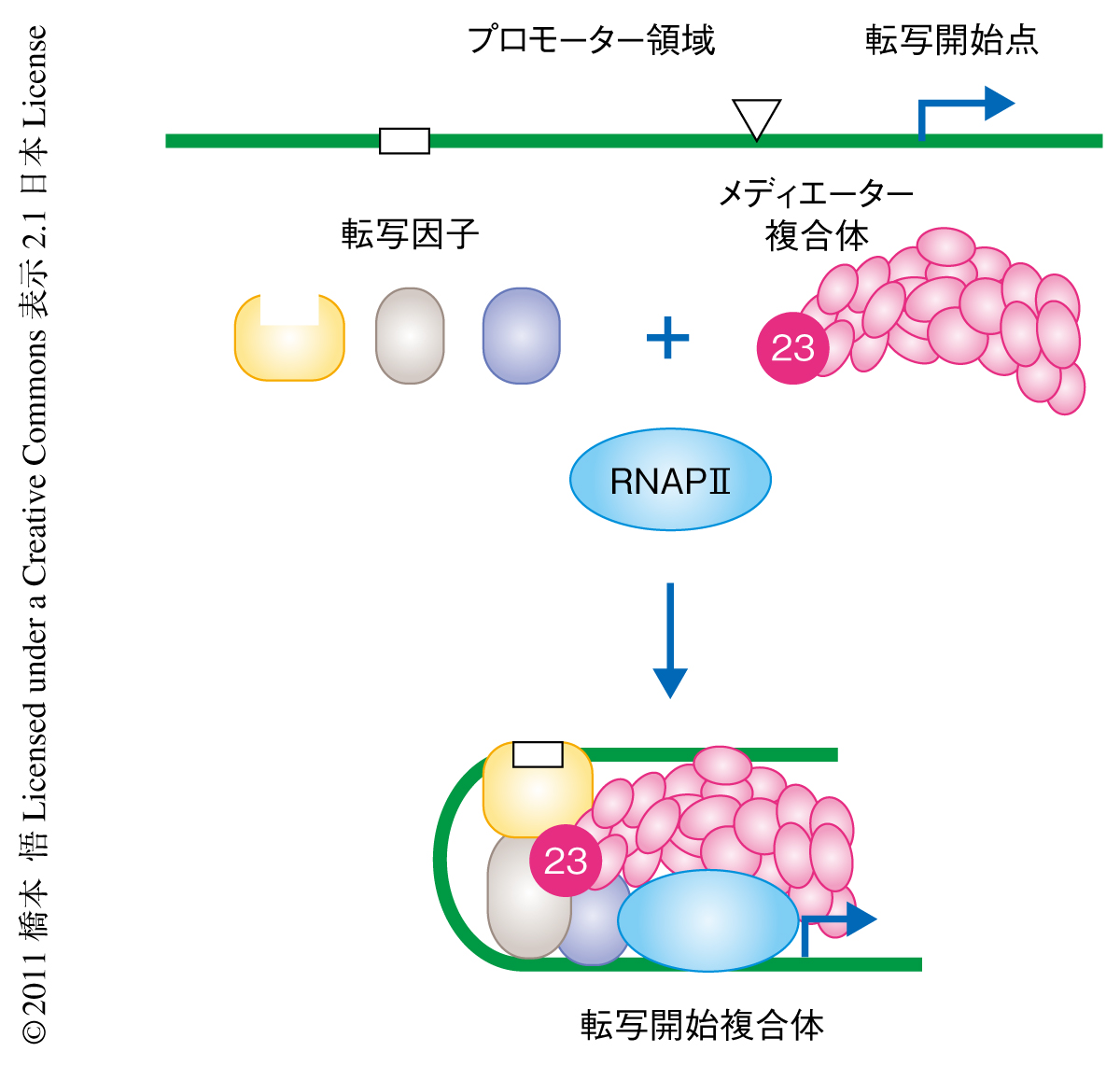
メディエーター複合体は20以上にものぼる多数のサブユニットから構成され,さらに,ヘッド,ミドル,テイル,キナーゼとよばれる4つのモジュールに分けられる2).しかしながら,その複雑さおよび巨大さのため,詳細な分子機構は十分には解明されていない.このため,メディエーター複合体のサブユニットに変異をもつ疾患の解析は,転写調節機構の解明に大きく役立つことが期待される.今回,筆者らは,MED23遺伝子の変異を知的障害者の家系から同定することにより,メディエーター複合体の分子機構の一部を解明するとともに,知的障害と即時発現型遺伝子における発現調節の異常との関係性を見い出した.
同意のもと,無症候性の知的障害を呈するアルジェリアの近親婚家系に対し連鎖解析法などを用いた遺伝学的な解析を行ったところ,MED23遺伝子の変異(1850番目のGがAに変異することで,Arg617がGlnに変異)を同定することに成功した.この変異は一塩基多型ではなく,また,Arg617は高等生物において広く保存されていることがわかった.つづいて,患者の皮膚組織からの線維芽細胞の培養を確立し,以後,この培養細胞を用いてさまざまな分子生物学的な実験を行った.変異型MED23の発現量は野生型MED23と同等であり,免疫沈降法を用いた実験によりメディエーター複合体の形成には影響をあたえていないことが示された.
マウスES細胞ではMED23が血清刺激ののちの即時発現型遺伝子の発現に関与しているとの報告がある3).そこで,血清刺激の前後の細胞より抽出したRNAを用いてマイクロアレイ解析を行ったところ,野生型の細胞にくらべ変異型MED23をもつ細胞ではいくつかの即時発現型遺伝子において発現異常を認めた.そのなかからまったく逆の発現パターンを示す2つの遺伝子,JUN遺伝子(発現なし)とFOS遺伝子(過剰発現)に注目し詳細な解析をつづけることにした.
血清刺激ののちのJUN遺伝子およびFOS遺伝子の発現量を確認したところ,マイクロアレイ解析の結果と同様に,変異型MED23をもつ細胞ではJUN遺伝子はまったく発現せず,逆に,FOS遺伝子は過剰発現していた.これら変異型MED23をもつ細胞における遺伝子発現の異常は,野生型MED23を発現するプラスミドを導入することにより野生型の細胞と同等のレベルに改善された.JUN遺伝子は血清刺激のほかにも紫外線などの外的な因子により誘導されるが,紫外線を照射した変異型MED23をもつ細胞には,野生型の細胞と同様にJUN遺伝子の発現がみられた.さらに,ホルモン応答遺伝子の発現量を確認したが,野生型の細胞と変異型MED23をもつ細胞とのあいだに差は認められなかった.以上より,変異型MED23はある特定の遺伝子に対し特定の条件での発現にのみ影響を及ぼすことがわかった.
つぎに,JUN遺伝子およびFOS遺伝子のプロモーター領域において,実際に転写開始複合体が正常に形成されているかどうかを確認するためクロマチン免疫沈降法を行った.変異型MED23をもつ細胞のJUN遺伝子プロモーター領域では,血清刺激ののちメディエーター複合体や基本転写因子であるTFIIHは誘導されていなかったが,RNAポリメラーゼIIは誘導されていた.TFIIHはRNAポリメラーゼIIのリン酸化キナーゼであり,このリン酸化は転写開始に必須である.また,転写伸長に不可欠なもうひとつのRNAポリメラーゼIIのリン酸化キナーゼあるP-TEFbも,変異型MED23をもつ細胞では誘導されていなかった.一方,FOS遺伝子プロモーター領域では,RNAポリメラーゼII,メディエーター複合体,TFIIH,P-TEFbのすべてが野生型の細胞と変わらず誘導されていた.
JUN遺伝子のプロモーター領域の上流にはTCF4結合領域があり,TCF4およびその下流にAP1が結合することによりDNAループ構造が形成され遺伝子発現の開始することが知られている4).変異型MED23をもつ細胞ではクロマチン免疫沈降法によりJUN遺伝子プロモーター領域においてAP1の局在が認められたが,TCF4の誘導はみられず,3C(chromatin conformation capture)法によっても血清刺激によるクロマチン構造の変化は認められなかった.さらに,野生型の細胞ではMED23がTCF4と複合体を形成していることも確認された.これらのことより,メディエーター複合体がJUN遺伝子プロモーター領域の立体構造の変化に関与していることが明らかになった.
FOS遺伝子のプロモーター領域にはELK1結合領域があり,血清刺激により誘導されたELK1がMED23と結合することによりFOS遺伝子の発現は活性化される5).しかしながら,ELK1結合領域にはELK1のパラログであるELK3も結合することができ,変異型MED23をもつ細胞では野生型の細胞とは異なりELK3の誘導が顕著に増加していた.本来,ELK1とELK3は同じ領域において競合しているが,野生型の細胞ではELK1が,変異型MED23をもつ細胞ではELK3が,優位に局在することによりFOS遺伝子の発現量に違いが生じたものと考えられた.
MED23のほかにも,MED12や基本転写因子TFIIHの変異により知的障害の生じることが知られている6,7).これらの変異をもつ細胞を用いて血清刺激ののちの即時発現型遺伝子の発現を定量したところ,予想どおり,その発現調節に異常が認められた.また,MED23の変異とは異なり,MED12やTFIIHの変異では即時発現型遺伝子のほかにもさまざまなホルモン応答遺伝子において発現異常がみられたが,これらの変異では臨床症状においても知的障害のほかにさまざまな全身所見が認められる.これらのことより,即時発現型遺伝子の適切な発現が認知機能の形成に必要であることが示唆された.
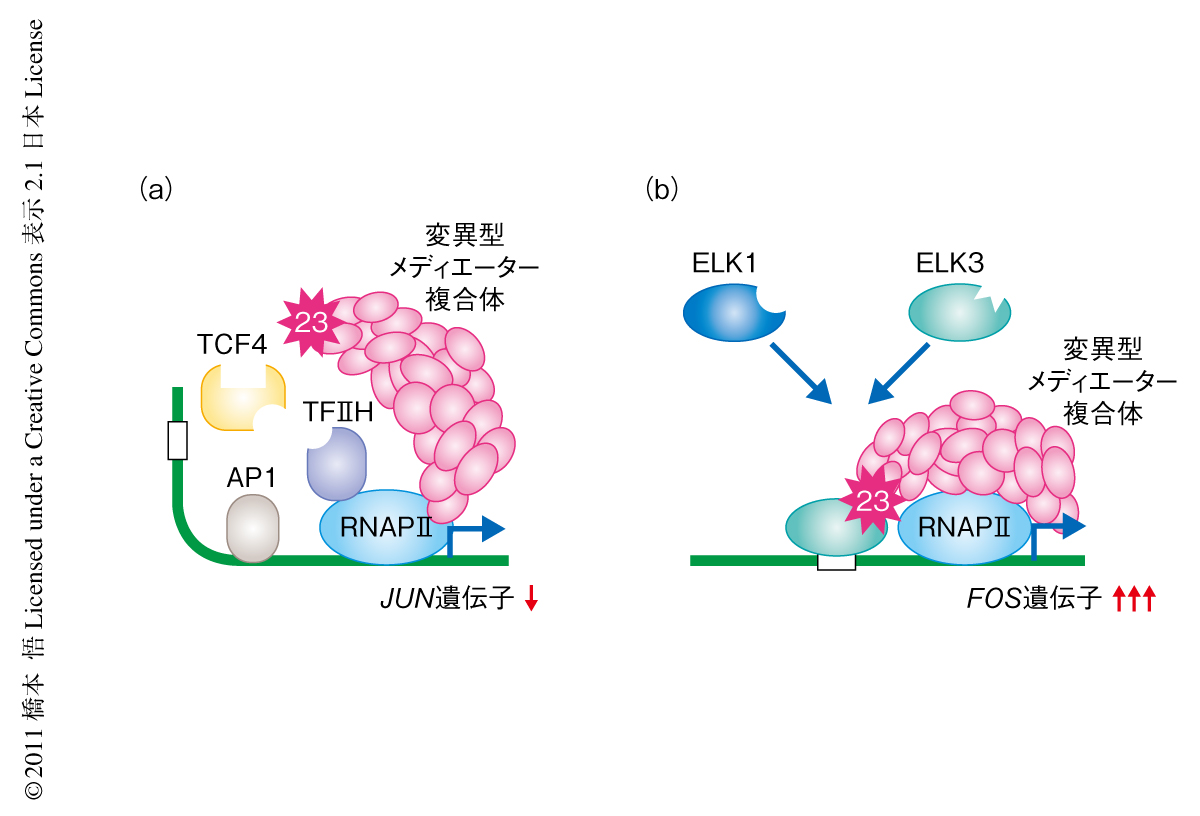
MED23遺伝子において新たに見い出した変異では,血清刺激ののちの即時発現型遺伝子の発現調節の異常を認めた.JUN遺伝子においては,変異型MED23によりメディエーター複合体がプロモーター領域に誘導されず,同時に,さまざまな転写因子の誘導が阻害されることによりクロマチンに構造変化が生じないため,結果として,JUN遺伝子の発現は抑制されていた(図2a).FOS遺伝子においては,変異型MED23によりメディエーター複合体は本来の結合相手であるELK1との結合が阻害され,代わりの結合相手であるELK3によりFOS遺伝子の発現は過剰になっていた(図2b).変異型MED23をもつ細胞ではこれら相反する発現調節の異常を示していたが,どちらも遺伝子プロモーター領域における複雑なタンパク質ネットワークの破綻に起因していることが明らかになった.
筆者らは,この例のほかにも,色素性乾皮症や硫黄欠乏性毛髪発育異常症などにおいて遺伝子発現の調節異常と病態との関連性を明らかにしてきており,これらの疾患を総じ“転写異常症候群”(transcription syndrome)という概念を提唱している.
略歴:2008年 熊本大学大学院医学教育部博士課程 修了,同年よりフランスInstitute of Genetics and Molecular and Cellular Biology博士研究員.
研究テーマ:色素性乾皮症およびその類縁疾患の病態解析.
抱負:人生を捧げる研究ではなく,人生を彩る研究を!
© 2011 橋本 悟 Licensed under CC 表示 2.1 日本
(フランスInstitute of Genetics and Molecular and Cellular Biology,Program of Functional Genomics & Cancer)
email:橋本 悟
DOI: 10.7875/first.author.2011.135
MED23 mutation links intellectual disability to dysregulation of immediate early gene expression.
Satoru Hashimoto, Sarah Boissel, Mohammed Zarhrate, Marlène Rio, Arnold Munnich, Jean-Marc Egly, Laurence Colleaux
Science, 333, 1161-1163 (2011)
要 約
MED23は遺伝子発現において重要な役割を担うメディエーター複合体のサブユニットのひとつである.筆者らは,常染色体劣勢の遺伝様式を示す知的障害者の家系より,新規にMED23遺伝子の変異を同定した.この変異では転写因子からの情報をRNAポリメラーゼIIに伝達するというメディエーター複合体の機能が阻害されており,血清による刺激ののちのJUN遺伝子およびFOS遺伝子を含む一部の即時発現型遺伝子の発現調節に異常を認めた.さらに,これらの即時発現型遺伝子における発現調節の異常は,知的障害をきたすことの知られる,ほかのメディエーター複合体のサブユニットや基本転写因子の変異においても確認された.これらのことは,中枢神経系の発達においてメディエーター複合体が重要であることを示すとともに,即時発現型遺伝子における発現調節の異常が認知障害の指標となりうることを示唆した.
はじめに
真核生物においてmRNAはRNAポリメラーゼIIのみにより合成(転写)されるが,その過程は多くの転写因子により制御されている.細胞の内外に生じるさまざまな刺激は転写因子を活性化させ,転写因子は遺伝子の上流のプロモーター領域にある特異的な配列に結合する.結合した転写因子はその下流に位置するRNAポリメラーゼIIおよび付属する基本転写因子に刺激を伝達するが,この刺激の伝達により転写の開始することが知られている.メディエーター複合体は転写因子とRNAポリメラーゼIIとのあいだの架け橋としてはたらき,多くの転写因子と結合することにより巨大なタンパク質ネットワークを形成することで複雑な転写調節が行われている1)(図1).
メディエーター複合体は20以上にものぼる多数のサブユニットから構成され,さらに,ヘッド,ミドル,テイル,キナーゼとよばれる4つのモジュールに分けられる2).しかしながら,その複雑さおよび巨大さのため,詳細な分子機構は十分には解明されていない.このため,メディエーター複合体のサブユニットに変異をもつ疾患の解析は,転写調節機構の解明に大きく役立つことが期待される.今回,筆者らは,MED23遺伝子の変異を知的障害者の家系から同定することにより,メディエーター複合体の分子機構の一部を解明するとともに,知的障害と即時発現型遺伝子における発現調節の異常との関係性を見い出した.
1.MED23遺伝子における変異の同定
同意のもと,無症候性の知的障害を呈するアルジェリアの近親婚家系に対し連鎖解析法などを用いた遺伝学的な解析を行ったところ,MED23遺伝子の変異(1850番目のGがAに変異することで,Arg617がGlnに変異)を同定することに成功した.この変異は一塩基多型ではなく,また,Arg617は高等生物において広く保存されていることがわかった.つづいて,患者の皮膚組織からの線維芽細胞の培養を確立し,以後,この培養細胞を用いてさまざまな分子生物学的な実験を行った.変異型MED23の発現量は野生型MED23と同等であり,免疫沈降法を用いた実験によりメディエーター複合体の形成には影響をあたえていないことが示された.
2.即時発現型遺伝子の発現パターン
マウスES細胞ではMED23が血清刺激ののちの即時発現型遺伝子の発現に関与しているとの報告がある3).そこで,血清刺激の前後の細胞より抽出したRNAを用いてマイクロアレイ解析を行ったところ,野生型の細胞にくらべ変異型MED23をもつ細胞ではいくつかの即時発現型遺伝子において発現異常を認めた.そのなかからまったく逆の発現パターンを示す2つの遺伝子,JUN遺伝子(発現なし)とFOS遺伝子(過剰発現)に注目し詳細な解析をつづけることにした.
血清刺激ののちのJUN遺伝子およびFOS遺伝子の発現量を確認したところ,マイクロアレイ解析の結果と同様に,変異型MED23をもつ細胞ではJUN遺伝子はまったく発現せず,逆に,FOS遺伝子は過剰発現していた.これら変異型MED23をもつ細胞における遺伝子発現の異常は,野生型MED23を発現するプラスミドを導入することにより野生型の細胞と同等のレベルに改善された.JUN遺伝子は血清刺激のほかにも紫外線などの外的な因子により誘導されるが,紫外線を照射した変異型MED23をもつ細胞には,野生型の細胞と同様にJUN遺伝子の発現がみられた.さらに,ホルモン応答遺伝子の発現量を確認したが,野生型の細胞と変異型MED23をもつ細胞とのあいだに差は認められなかった.以上より,変異型MED23はある特定の遺伝子に対し特定の条件での発現にのみ影響を及ぼすことがわかった.
3.転写開始複合体の形成の確認
つぎに,JUN遺伝子およびFOS遺伝子のプロモーター領域において,実際に転写開始複合体が正常に形成されているかどうかを確認するためクロマチン免疫沈降法を行った.変異型MED23をもつ細胞のJUN遺伝子プロモーター領域では,血清刺激ののちメディエーター複合体や基本転写因子であるTFIIHは誘導されていなかったが,RNAポリメラーゼIIは誘導されていた.TFIIHはRNAポリメラーゼIIのリン酸化キナーゼであり,このリン酸化は転写開始に必須である.また,転写伸長に不可欠なもうひとつのRNAポリメラーゼIIのリン酸化キナーゼあるP-TEFbも,変異型MED23をもつ細胞では誘導されていなかった.一方,FOS遺伝子プロモーター領域では,RNAポリメラーゼII,メディエーター複合体,TFIIH,P-TEFbのすべてが野生型の細胞と変わらず誘導されていた.
4.JUN遺伝子プロモーター領域における立体構造の変化
JUN遺伝子のプロモーター領域の上流にはTCF4結合領域があり,TCF4およびその下流にAP1が結合することによりDNAループ構造が形成され遺伝子発現の開始することが知られている4).変異型MED23をもつ細胞ではクロマチン免疫沈降法によりJUN遺伝子プロモーター領域においてAP1の局在が認められたが,TCF4の誘導はみられず,3C(chromatin conformation capture)法によっても血清刺激によるクロマチン構造の変化は認められなかった.さらに,野生型の細胞ではMED23がTCF4と複合体を形成していることも確認された.これらのことより,メディエーター複合体がJUN遺伝子プロモーター領域の立体構造の変化に関与していることが明らかになった.
5.FOS遺伝子プロモーター領域における転写因子の競合
FOS遺伝子のプロモーター領域にはELK1結合領域があり,血清刺激により誘導されたELK1がMED23と結合することによりFOS遺伝子の発現は活性化される5).しかしながら,ELK1結合領域にはELK1のパラログであるELK3も結合することができ,変異型MED23をもつ細胞では野生型の細胞とは異なりELK3の誘導が顕著に増加していた.本来,ELK1とELK3は同じ領域において競合しているが,野生型の細胞ではELK1が,変異型MED23をもつ細胞ではELK3が,優位に局在することによりFOS遺伝子の発現量に違いが生じたものと考えられた.
6.即時発現型遺伝子における発現調節の異常と知的障害をともなうほかの遺伝子変異
MED23のほかにも,MED12や基本転写因子TFIIHの変異により知的障害の生じることが知られている6,7).これらの変異をもつ細胞を用いて血清刺激ののちの即時発現型遺伝子の発現を定量したところ,予想どおり,その発現調節に異常が認められた.また,MED23の変異とは異なり,MED12やTFIIHの変異では即時発現型遺伝子のほかにもさまざまなホルモン応答遺伝子において発現異常がみられたが,これらの変異では臨床症状においても知的障害のほかにさまざまな全身所見が認められる.これらのことより,即時発現型遺伝子の適切な発現が認知機能の形成に必要であることが示唆された.
おわりに
MED23遺伝子において新たに見い出した変異では,血清刺激ののちの即時発現型遺伝子の発現調節の異常を認めた.JUN遺伝子においては,変異型MED23によりメディエーター複合体がプロモーター領域に誘導されず,同時に,さまざまな転写因子の誘導が阻害されることによりクロマチンに構造変化が生じないため,結果として,JUN遺伝子の発現は抑制されていた(図2a).FOS遺伝子においては,変異型MED23によりメディエーター複合体は本来の結合相手であるELK1との結合が阻害され,代わりの結合相手であるELK3によりFOS遺伝子の発現は過剰になっていた(図2b).変異型MED23をもつ細胞ではこれら相反する発現調節の異常を示していたが,どちらも遺伝子プロモーター領域における複雑なタンパク質ネットワークの破綻に起因していることが明らかになった.
筆者らは,この例のほかにも,色素性乾皮症や硫黄欠乏性毛髪発育異常症などにおいて遺伝子発現の調節異常と病態との関連性を明らかにしてきており,これらの疾患を総じ“転写異常症候群”(transcription syndrome)という概念を提唱している.
文 献
- Malik, S. & Roeder, R. G.: The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet., 11, 761-772 (2010)[PubMed]
- Casamassimi, A. & Napoli, C.: Mediator complexes and eukaryotic transcription regulation: an overview. Biochimie, 89, 1439-1446 (2007)[PubMed]
- Stevens, J. L., Cantin, G. T., Wang, G. et al.: Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science, 296, 755-758 (2002)[PubMed]
- Nateri, A. S., Spencer-Dene, B. & Behrens, A.: Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature, 437, 281-285 (2005)[PubMed]
- Buchwalter, G., Gross, C. & Wasylyk, B.: Ets ternary complex transcription factors. Gene, 324, 1-14 (2004)[PubMed]
- Risheg, H., Graham, J. M. Jr, Clark, R. D. et al.: A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet, 39, 451-453 (2007)[PubMed]
- Cleaver, J. E., Lam, E. T. & Revet, I.: Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat. Rev. Genet., 10, 756-768 (2009)[PubMed]
著者プロフィール
略歴:2008年 熊本大学大学院医学教育部博士課程 修了,同年よりフランスInstitute of Genetics and Molecular and Cellular Biology博士研究員.
研究テーマ:色素性乾皮症およびその類縁疾患の病態解析.
抱負:人生を捧げる研究ではなく,人生を彩る研究を!
© 2011 橋本 悟 Licensed under CC 表示 2.1 日本
