休眠複製開始点はS期で停止した複製フォークの蓄積を防ぎ染色体の不分離とがんの形成を抑制する
川端 剛・島 直子
(米国Minnesota大学Department of Genetics, Cell Biology and Development)
email:島 直子
DOI: 10.7875/first.author.2011.057
Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression.
Tsuyoshi Kawabata, Spencer W. Luebben, Satoru Yamaguchi, Ivar Ilves, Ilze Matise, Tavanna Buske, Michael R. Botchan, Naoko Shima
Molecular Cell, 41, 543-553 (2011)
ゲノム情報を正確に維持するためDNA複製は細胞周期あたり1回だけ行われる.真核生物では実際に複製がはじまる領域の数よりも多数の潜在的な複製開始点が存在し,これら余剰な複製開始点(休眠複製開始点)は複製が阻害された状況にあっても複製を完了するためのバックアップとして機能する.しかしながら,休眠複製開始点がゲノム安定性を維持する具体的なしくみ,また,発がんとの関連は不明であった.今回,筆者らは,休眠複製開始点が通常のDNA複製において局所的に停止した複製フォークをレスキューし,不完全な複製により生じる染色体の不分離を抑制することでがんの形成を防いでいることを明らかにした.
すべての遺伝情報を2つの娘細胞に受け継ぐため,細胞分裂を行う細胞は細胞周期のS期においてゲノムを正確に複製しなければならない.原核生物である大腸菌のゲノムサイズは4600 kbであり,複製開始点が1つしかないにもかかわらず複製は約40分間で完了する.しかし,真核生物のゲノム,とくにヒトなどの哺乳類のゲノムははるかに巨大であり,複製開始点が1つだけではゲノムを複製するのに非常に長い時間のかかる計算になるが,真核生物は1つの染色体に複数の複製開始点をもつことでこの問題を解決している.この事実は同時に,真核生物においてDNA複製の開始が厳密に制御されなくてはならないことを示している.つまり,いつ,どこに,どれだけ,複製開始点が形成され,それらがS期でどのように活性化されるかを制御する機構が存在している.
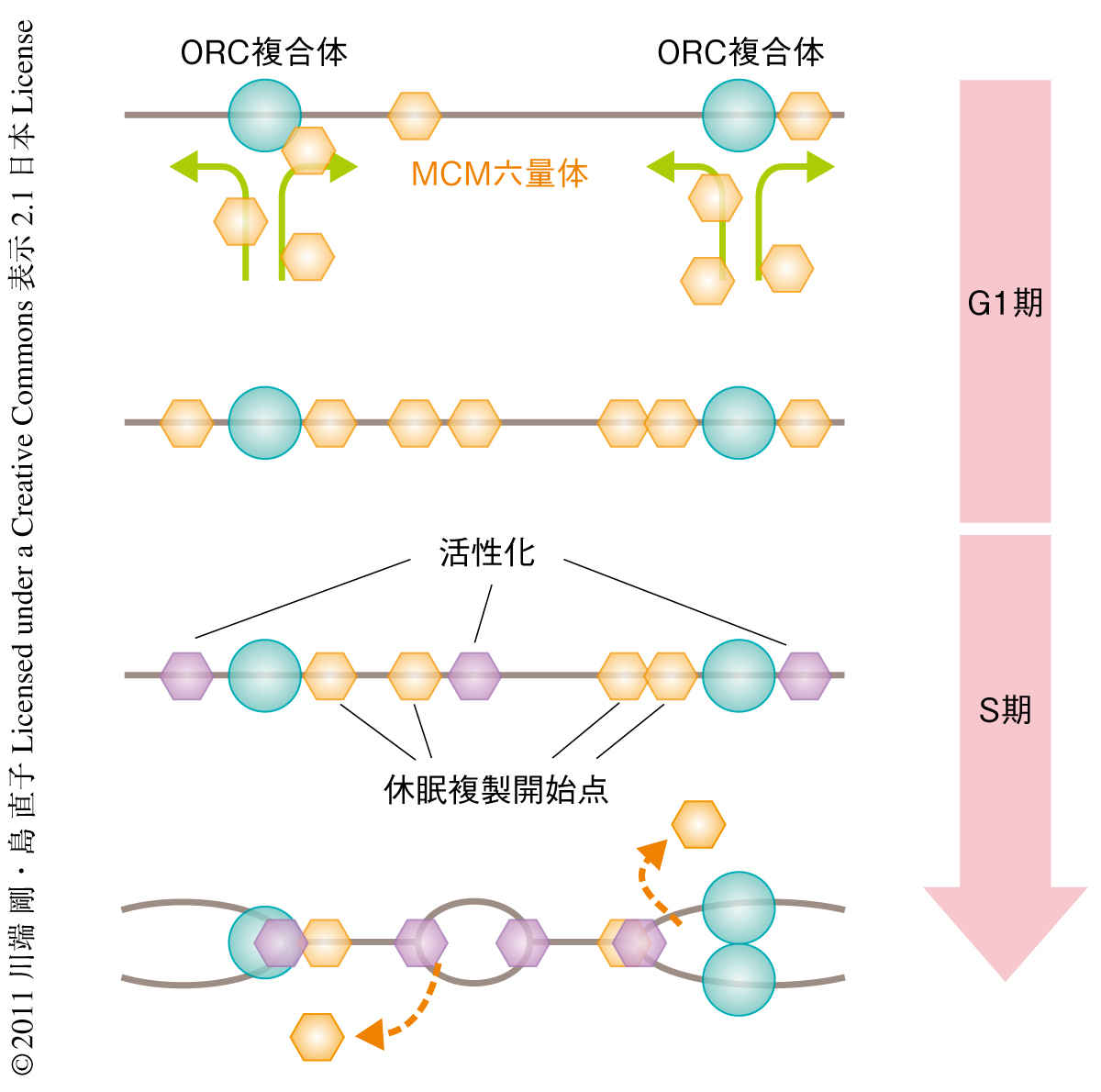
DNA複製開始点は,MCM2~MCM7からなるDNA複製ヘリカーゼであるMCM六量体が,ORC1~ORC6からなるORC複合体によりゲノムに結合することで形成される(図1).これをDNA複製のライセンシングとよぶ1).この過程は細胞周期のG1期のみに起こりS期とG2期では抑制されている.これはゲノムが部分的に重複して複製されることを防ぐためである.真核生物ではG1期において,S期で実際に活性化される数よりも多くの複製開始点がすでに準備されており,緊急事態ではこれらの休眠複製開始点(dormant replication origin)が活性化され停止した複製フォークを補い複製を完了する2,3)(図2).

MCM六量体の構成タンパク質のノックアウトマウスは胎生致死となるため解析が困難であるが,筆者らの研究室では,以前,N-エチル-N-ニトロソ尿素を用いた突然変異誘発スクリーニングによりMcm4低形質アレルをもつMcm4Chaos3変異マウスを同定した4).この変異は点突然変異でありMCM4の345番目のPheがIleに置換している.Mcm4Chaos3ホモ変異マウスでは平均12カ月で乳がんがみられる.Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞ではMCM六量体の構成タンパク質のうちMCM4,MCM2およびMCM7のタンパク質量の低下がみられたため休眠複製開始点の低下はがんの形成を促進するものと予想されたが,外因性のストレスがない条件において休眠複製開始点がどのようにがんを抑制しているかは謎であった.今回の報告で,筆者らは,休眠複製開始点は通常のDNA複製においても停止した複製フォークの蓄積を防ぎ,さらに部分的に複製されないゲノム領域の生成を防ぐことで,染色体の正確な分離を保証していることを明らかにした.
今回,実際にMcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞で休眠複製開始点の数が低下していることを示された.半定量的ウェスタンブロットにより,MCM六量体を構成するMCM2~MCM7すべてのクロマチン画分への結合がおおよそ50%低下していることがわかった.また,ビオチン-dUTPの取り込みにより複製されたDNAを標識して可視化するDNAファイバー解析5) を行ったところ,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞において通常の条件で活性化された複製開始点の密度は野生型細胞と有意な差はみられなかった.しかしながら,DNA複製にはたらくDNAポリメラーゼα,DNAポリメラーゼδおよびDNAポリメラーゼεの阻害剤であるアフィディコリンによる処理が野生型細胞における複製開始点の密度を有意に上昇させた(休眠複製開始点が活性化)のに対し,Mcm4Chaos3ホモ変異細胞では複製開始点の密度上昇はアフィディコリン未処理に比べてやや上昇するものの野生型細胞のおおよそ半分であった.これより,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞では休眠複製開始点の数が半分ほどに低下していると結論づけた.
以前の仮説では,この休眠複製開始点の低下は通常のDNA複製には影響をあたえないと考えられていた.しかしながら,DNA複製の双方向への伸張をDNAファイバー解析によりみたところ,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞では片方の短い非対称のDNA複製が野生型細胞よりも有意に多くみられた.さらに,停止した複製フォークのマーカーであるRPAおよびリン酸化RAD17のフォーカス形成がMcm4Chaos3ホモ変異細胞では多くみられた.これより,休眠複製開始点は通常のDNA複製においても局所的な複製フォークの停止を補完する機能のあることが示された.
MCM六量体はほかに2つの補因子CDC45およびGINSと結合してCMG複合体を形成することでDNAヘリカーゼとして機能する6).キイロショウジョウバエでChaos3変異MCM4を含むCMG複合体を構築しヘリカーゼ活性をin vitroで解析した.変異型CMG複合体のおのおのの構成タンパク質は野生型CMG複合体と同じ量比で精製されたが,その収量は少なかった.CMG複合体に含まれるMCM2~MCM7をMonoQカラムで分画したところ,変異型CMG複合体は野生型CMG複合体にはみられない低分子量の複合体と野生型と同じサイズの複合体とに分かれることがわかった.これより,Mcm4Chaos3変異はMCM六量体の形成を阻害するが,補因子と結合してCMG複合体が形成されたあとでは安定化するものと予想された.さらに,変異型CMG複合体が野生型CMG複合体と同じ程度もしくはやや高いヘリカーゼ活性を示すことをふまえると,Mcm4Chaos3変異細胞の表現型はヘリカーゼ活性の低下ではなく休眠複製開始点の数が低下したことによりひき起こされているものと考えられた.
Mcm4Chaos3ホモ変異細胞において停止した複製フォークの蓄積していることがわかったが,これらは相同組換え修復によりある程度まで補われることが予想されていた7).RAD51は相同組換え修復における主要なタンパク質であるが,このRAD51のフォーカス形成がMcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞で上昇していた.このことから休眠複製開始点の低下は相同組換え修復を活性化するものと予想されたが,また同時に,リコンビナーゼ阻害タンパク質として機能するBLMのフォーカス形成も上昇していた.そこで,実際の相同組換え修復の頻度をFYDR(fluorescent yellow direct repeat)法8) により測定した.Mcm4Chaos3ホモ変異マウスの2週齢胚に自然発生する相同組換え修復の頻度は野生型マウスよりもやや高かったが統計的に有意な差はみられなかった.このことからFYDR法により検出できる相同組換え修復はBLMにより抑制されているものと考えられた.しかしながら,以前の報告によると,RAD51による複製フォークのレスキューはFYDR法に類似した方法では検出できないことが示されており9)(遺伝子変換と姉妹染色分体の交換をともなわない正確なレスキューは検出できないためと予想される),依然としてRAD51のフォーカス形成にともなうレスキューが増加している可能性はある.
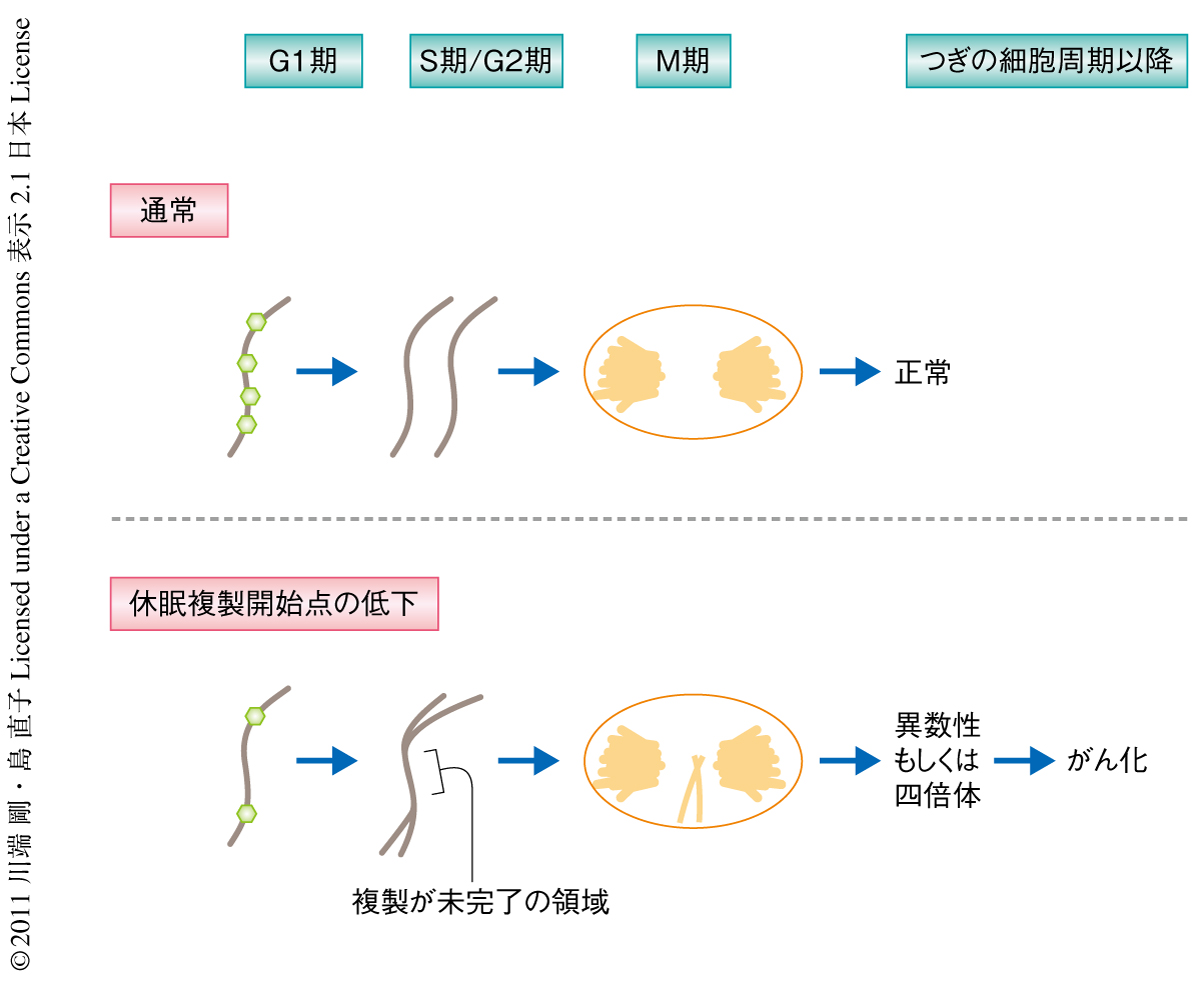
不完全なDNA複製を示すマーカーとして,Fanconi貧血症の原因遺伝子のひとつによりコードされるFANCD2のフォーカス形成が報告されている10).Mcm4Chaos3ホモ変異細胞では外因性のDNA損傷がない条件でM期においてFANCD2の姉妹フォーカス形成が野生型細胞の4倍以上に上昇していた.このことからMcm4Chaos3ホモ変異細胞ではRAD51経路およびBLM経路が活性化されているにもかかわらずDNA複製は不完全となることが示唆された.FANCD2のフォーカス形成はM期のおわりまでもちこされており,これと一致して,一部の染色体分離の遅れがみられた.G1期に入ると,分離できなかった染色体の形成する小核の頻度が野生型細胞の2倍ほど観察された.さらに,この小核のおよそ4割が2本鎖DNA切断のマーカーであるリン酸化ヒストンH2AXに対する抗体で,また,およそ6割がセントロメアのマーカーであるCENP-Aに対する抗体で染色された.これは,染色体の断片もしくは染色体が核から失われていることを示唆していた(図3).この事実は蛍光in situハイブリダイゼーションによる染色体数の変化によっても確認された.
以前の報告で,C3HeB/FeJ(C3H)系統のMcm4Chaos3ホモ変異マウスは乳がんに高い感受性を示したが,ほかの系統でも同様であるかどうかは不明であった.そこで,B6(C57BL/6J)系統およびC3H/B6系統のMcm4Chaos3ホモ変異マウスを作製しがんの形成への影響をみた.C3H/B6系統の多くが組織球性肉腫に感受性を示し,F1においては組織球性肉腫とリンパ腫が大半をしめ,少数が乳がんを示した.また,がんを形成する平均期間はこれらのマウスで大きく変わらなかった.これより,休眠複製開始点の低下によるがん形成のスペクトラムは遺伝学的な背景の影響を強くうけることが示された.
これまでにも,停止した複製フォークが付近の複製開始点からの複製フォークによってレスキューされる可能性は指摘されていたが,その重要性はみすごされがちであった.今回,筆者らは,通常のDNA複製において停止した複製フォークをレスキューするため休眠複製開始点の欠かせないことを示した.今後,休眠複製開始点によるレスキューと相同組換え修復あるいはほかの経路との詳細な関係を調べ,それらががん化を抑制するしくみを解明する予定である.
略歴:2007年 埼玉大学大学院理工学研究科博士課程 修了,同年より米国Minnesota大学 研究員.
研究テーマ:停止した複製フォークとゲノム安定性との関係.
関心事:休眠複製開始点の役割にフォーカスしています.
島 直子(Naoko Shima)
米国Minnesota大学Assistant Professor.
© 2011 川端 剛・島 直子 Licensed under CC 表示 2.1 日本
(米国Minnesota大学Department of Genetics, Cell Biology and Development)
email:島 直子
DOI: 10.7875/first.author.2011.057
Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression.
Tsuyoshi Kawabata, Spencer W. Luebben, Satoru Yamaguchi, Ivar Ilves, Ilze Matise, Tavanna Buske, Michael R. Botchan, Naoko Shima
Molecular Cell, 41, 543-553 (2011)
要 約
ゲノム情報を正確に維持するためDNA複製は細胞周期あたり1回だけ行われる.真核生物では実際に複製がはじまる領域の数よりも多数の潜在的な複製開始点が存在し,これら余剰な複製開始点(休眠複製開始点)は複製が阻害された状況にあっても複製を完了するためのバックアップとして機能する.しかしながら,休眠複製開始点がゲノム安定性を維持する具体的なしくみ,また,発がんとの関連は不明であった.今回,筆者らは,休眠複製開始点が通常のDNA複製において局所的に停止した複製フォークをレスキューし,不完全な複製により生じる染色体の不分離を抑制することでがんの形成を防いでいることを明らかにした.
はじめに
すべての遺伝情報を2つの娘細胞に受け継ぐため,細胞分裂を行う細胞は細胞周期のS期においてゲノムを正確に複製しなければならない.原核生物である大腸菌のゲノムサイズは4600 kbであり,複製開始点が1つしかないにもかかわらず複製は約40分間で完了する.しかし,真核生物のゲノム,とくにヒトなどの哺乳類のゲノムははるかに巨大であり,複製開始点が1つだけではゲノムを複製するのに非常に長い時間のかかる計算になるが,真核生物は1つの染色体に複数の複製開始点をもつことでこの問題を解決している.この事実は同時に,真核生物においてDNA複製の開始が厳密に制御されなくてはならないことを示している.つまり,いつ,どこに,どれだけ,複製開始点が形成され,それらがS期でどのように活性化されるかを制御する機構が存在している.
DNA複製開始点は,MCM2~MCM7からなるDNA複製ヘリカーゼであるMCM六量体が,ORC1~ORC6からなるORC複合体によりゲノムに結合することで形成される(図1).これをDNA複製のライセンシングとよぶ1).この過程は細胞周期のG1期のみに起こりS期とG2期では抑制されている.これはゲノムが部分的に重複して複製されることを防ぐためである.真核生物ではG1期において,S期で実際に活性化される数よりも多くの複製開始点がすでに準備されており,緊急事態ではこれらの休眠複製開始点(dormant replication origin)が活性化され停止した複製フォークを補い複製を完了する2,3)(図2).
MCM六量体の構成タンパク質のノックアウトマウスは胎生致死となるため解析が困難であるが,筆者らの研究室では,以前,N-エチル-N-ニトロソ尿素を用いた突然変異誘発スクリーニングによりMcm4低形質アレルをもつMcm4Chaos3変異マウスを同定した4).この変異は点突然変異でありMCM4の345番目のPheがIleに置換している.Mcm4Chaos3ホモ変異マウスでは平均12カ月で乳がんがみられる.Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞ではMCM六量体の構成タンパク質のうちMCM4,MCM2およびMCM7のタンパク質量の低下がみられたため休眠複製開始点の低下はがんの形成を促進するものと予想されたが,外因性のストレスがない条件において休眠複製開始点がどのようにがんを抑制しているかは謎であった.今回の報告で,筆者らは,休眠複製開始点は通常のDNA複製においても停止した複製フォークの蓄積を防ぎ,さらに部分的に複製されないゲノム領域の生成を防ぐことで,染色体の正確な分離を保証していることを明らかにした.
1.Mcm4Chaos3変異細胞では休眠複製開始点の数が低下している
今回,実際にMcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞で休眠複製開始点の数が低下していることを示された.半定量的ウェスタンブロットにより,MCM六量体を構成するMCM2~MCM7すべてのクロマチン画分への結合がおおよそ50%低下していることがわかった.また,ビオチン-dUTPの取り込みにより複製されたDNAを標識して可視化するDNAファイバー解析5) を行ったところ,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞において通常の条件で活性化された複製開始点の密度は野生型細胞と有意な差はみられなかった.しかしながら,DNA複製にはたらくDNAポリメラーゼα,DNAポリメラーゼδおよびDNAポリメラーゼεの阻害剤であるアフィディコリンによる処理が野生型細胞における複製開始点の密度を有意に上昇させた(休眠複製開始点が活性化)のに対し,Mcm4Chaos3ホモ変異細胞では複製開始点の密度上昇はアフィディコリン未処理に比べてやや上昇するものの野生型細胞のおおよそ半分であった.これより,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞では休眠複製開始点の数が半分ほどに低下していると結論づけた.
2.休眠複製開始点は通常のDNA複製においても役割をもつ
以前の仮説では,この休眠複製開始点の低下は通常のDNA複製には影響をあたえないと考えられていた.しかしながら,DNA複製の双方向への伸張をDNAファイバー解析によりみたところ,Mcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞では片方の短い非対称のDNA複製が野生型細胞よりも有意に多くみられた.さらに,停止した複製フォークのマーカーであるRPAおよびリン酸化RAD17のフォーカス形成がMcm4Chaos3ホモ変異細胞では多くみられた.これより,休眠複製開始点は通常のDNA複製においても局所的な複製フォークの停止を補完する機能のあることが示された.
3.Mcm4Chaos3変異はヘリカーゼ活性を低下させずに複合体の形成を阻害する
MCM六量体はほかに2つの補因子CDC45およびGINSと結合してCMG複合体を形成することでDNAヘリカーゼとして機能する6).キイロショウジョウバエでChaos3変異MCM4を含むCMG複合体を構築しヘリカーゼ活性をin vitroで解析した.変異型CMG複合体のおのおのの構成タンパク質は野生型CMG複合体と同じ量比で精製されたが,その収量は少なかった.CMG複合体に含まれるMCM2~MCM7をMonoQカラムで分画したところ,変異型CMG複合体は野生型CMG複合体にはみられない低分子量の複合体と野生型と同じサイズの複合体とに分かれることがわかった.これより,Mcm4Chaos3変異はMCM六量体の形成を阻害するが,補因子と結合してCMG複合体が形成されたあとでは安定化するものと予想された.さらに,変異型CMG複合体が野生型CMG複合体と同じ程度もしくはやや高いヘリカーゼ活性を示すことをふまえると,Mcm4Chaos3変異細胞の表現型はヘリカーゼ活性の低下ではなく休眠複製開始点の数が低下したことによりひき起こされているものと考えられた.
4.リコンビナーゼおよびリコンビナーゼ阻害タンパク質のフォーカス形成の上昇
Mcm4Chaos3ホモ変異細胞において停止した複製フォークの蓄積していることがわかったが,これらは相同組換え修復によりある程度まで補われることが予想されていた7).RAD51は相同組換え修復における主要なタンパク質であるが,このRAD51のフォーカス形成がMcm4Chaos3ホモ変異マウスに由来する胚性繊維芽細胞で上昇していた.このことから休眠複製開始点の低下は相同組換え修復を活性化するものと予想されたが,また同時に,リコンビナーゼ阻害タンパク質として機能するBLMのフォーカス形成も上昇していた.そこで,実際の相同組換え修復の頻度をFYDR(fluorescent yellow direct repeat)法8) により測定した.Mcm4Chaos3ホモ変異マウスの2週齢胚に自然発生する相同組換え修復の頻度は野生型マウスよりもやや高かったが統計的に有意な差はみられなかった.このことからFYDR法により検出できる相同組換え修復はBLMにより抑制されているものと考えられた.しかしながら,以前の報告によると,RAD51による複製フォークのレスキューはFYDR法に類似した方法では検出できないことが示されており9)(遺伝子変換と姉妹染色分体の交換をともなわない正確なレスキューは検出できないためと予想される),依然としてRAD51のフォーカス形成にともなうレスキューが増加している可能性はある.
5.DNA複製中間体はM期にもちこされ分裂後期に染色体の不分離をひき起こす
不完全なDNA複製を示すマーカーとして,Fanconi貧血症の原因遺伝子のひとつによりコードされるFANCD2のフォーカス形成が報告されている10).Mcm4Chaos3ホモ変異細胞では外因性のDNA損傷がない条件でM期においてFANCD2の姉妹フォーカス形成が野生型細胞の4倍以上に上昇していた.このことからMcm4Chaos3ホモ変異細胞ではRAD51経路およびBLM経路が活性化されているにもかかわらずDNA複製は不完全となることが示唆された.FANCD2のフォーカス形成はM期のおわりまでもちこされており,これと一致して,一部の染色体分離の遅れがみられた.G1期に入ると,分離できなかった染色体の形成する小核の頻度が野生型細胞の2倍ほど観察された.さらに,この小核のおよそ4割が2本鎖DNA切断のマーカーであるリン酸化ヒストンH2AXに対する抗体で,また,およそ6割がセントロメアのマーカーであるCENP-Aに対する抗体で染色された.これは,染色体の断片もしくは染色体が核から失われていることを示唆していた(図3).この事実は蛍光in situハイブリダイゼーションによる染色体数の変化によっても確認された.
6.Mcm4Chaos3変異は乳がん以外のがんも促進する
以前の報告で,C3HeB/FeJ(C3H)系統のMcm4Chaos3ホモ変異マウスは乳がんに高い感受性を示したが,ほかの系統でも同様であるかどうかは不明であった.そこで,B6(C57BL/6J)系統およびC3H/B6系統のMcm4Chaos3ホモ変異マウスを作製しがんの形成への影響をみた.C3H/B6系統の多くが組織球性肉腫に感受性を示し,F1においては組織球性肉腫とリンパ腫が大半をしめ,少数が乳がんを示した.また,がんを形成する平均期間はこれらのマウスで大きく変わらなかった.これより,休眠複製開始点の低下によるがん形成のスペクトラムは遺伝学的な背景の影響を強くうけることが示された.
おわりに
これまでにも,停止した複製フォークが付近の複製開始点からの複製フォークによってレスキューされる可能性は指摘されていたが,その重要性はみすごされがちであった.今回,筆者らは,通常のDNA複製において停止した複製フォークをレスキューするため休眠複製開始点の欠かせないことを示した.今後,休眠複製開始点によるレスキューと相同組換え修復あるいはほかの経路との詳細な関係を調べ,それらががん化を抑制するしくみを解明する予定である.
文 献
- Masai, H., Matsumoto, S. & You, Z.: Eukaryotic chromosome DNA replication: where, when, and how? Annu. Rev. Biochem., 79, 89-130 (2010)[PubMed]
- Ge, X. Q., Jackson, D. A. & Blow, J. J.: Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev., 21, 3331-3341 (2007)[PubMed]
- Ibarra, A., Schwob, E. & Mendez, J.: Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA, 105, 8956-8961 (2008)[PubMed]
- Shima, N., Alcaraz, A., Liachko, I. et al.: A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet., 39, 93-98 (2007)[PubMed]
- Sugimura, K., Takebayashi, S., Ogata, S. et al.: Non-denaturing fluorescence in situ hybridization to find replication origins in a specific genome region on the DNA fiber. Biosci. Biotechnol. Biochem., 71, 627-632 (2007)[PubMed]
- Ilves, I., Petojevic, T. & Pesavento, J. J.: Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell, 37, 247-258 (2010)[PubMed]
- Blow, J. J. & Gillespie, P. J.: Replication licensing and cancer: a fatal entanglement? Nat. Rev. Cancer, 8, 799-806 (2008)[PubMed]
- Hendricks, C. A., Almeida, K. H., Stitt, M. S. et al.: Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc. Natl. Acad. Sci. USA, 100, 6325-6330 (2003)[PubMed]
- Petermann, E., Orta, M. L., Issaeva, N. et al.: Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell, 37, 492-502 (2010)[PubMed]
- Chan, K. L., Palmai-Pallag, T., Ying, S. et al.: Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol., 11, 753-760 (2009)[PubMed]
著者プロフィール
略歴:2007年 埼玉大学大学院理工学研究科博士課程 修了,同年より米国Minnesota大学 研究員.
研究テーマ:停止した複製フォークとゲノム安定性との関係.
関心事:休眠複製開始点の役割にフォーカスしています.
島 直子(Naoko Shima)
米国Minnesota大学Assistant Professor.
© 2011 川端 剛・島 直子 Licensed under CC 表示 2.1 日本
