SIK2はTORC1-CREB経路を介してニューロンの障害を制御する
佐々木 勉1・竹森 洋2・北川一夫1
(1大阪大学大学院医学系研究科 神経内科学,2医薬基盤研究所創薬基盤研究部 代謝疾患関連タンパク探索プロジェクト)
email:佐々木 勉,北川一夫
DOI: 10.7875/first.author.2011.029
SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB.
Tsutomu Sasaki, Hiroshi Takemori, Yoshiki Yagita, Yasukazu Terasaki, Tatsuya Uebi, Nanao Horike, Hiroaki Takagi, Teruo Susumu, Hiroshi Teraoka, Ken-ichi Kusano, Osamu Hatano, Naoki Oyama, Yukio Sugiyama, Saburo Sakoda, Kazuo Kitagawa
Neuron, 69, 106-119 (2011)
転写因子CREBは脳に豊富に発現し,記憶,神経可塑性,細胞の生存など,幅広い神経機能あるいは病態に関与している.また,ニューロンにはSIK2およびTORC1が豊富に発現しており,SIK2はCREBを介した遺伝子発現を抑制している.筆者らは,大脳皮質のニューロンにおいて脳虚血の際にはCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVの活性化によりSIK2が分解され,TORC1-CREB経路を介して下流の神経保護因子の発現が誘導される,という新規のシグナル伝達経路を同定した.また,脳虚血の際のニューロンにおいてはNMDA型グルタミン酸受容体を介したCa2+の流入が重要であるが,とりわけNR2Aサブユニットをもつタイプがシナプス刺激における内因性の保護シグナルに関与していることを示した.また,SIK2ノックアウトマウスにおいてニューロンの生存の促進効果,脳梗塞サイズの縮小を認めた.今回の成果は,SIK2およびTORC1を標的とした治療が,脳梗塞のみならず認知症などを含めた神経変性疾患に対しても有効な治療につながりうる可能性を示唆した.
脳虚血は低酸素,酸化ストレス,炎症,グルタミン酸毒性などの複合的なストレスであり,これまでの研究により脳虚血の病態にかかわる多くの因子および経路が解明され多くの神経保護薬が検討されてきたが,臨床的にはいまだ十分であるとはいえない状況にある.とりわけ,虚血性のニューロン死をひき起こす細胞へのCa2+の流入経路としてNMDA型グルタミン酸受容体は重要なもののひとつであるが,MK-801をはじめとした汎グルタミン酸受容体拮抗薬は臨床的には有効な結果を得られなかった.NMDA型グルタミン酸受容体の活性化により下流の神経保護あるいは神経障害につながりうる多くの転写因子の活性がダイナミックに変化する1).とりわけ転写因子CREB(cAMP responsive element-binding protein)の活性化は,筆者らあるいはほかのグループも,重要な神経保護シグナルとして精力的に研究を行ってきた2,3).しかしながら,近年,グルタミン酸受容体のシナプスあるいはシナプス外での局在の差異あるいはサブタイプの違いなどの詳細な検討がなされ,それらの違いにより下流のシグナル伝達あるいは細胞の生死に決定的な違いのあることが報告された2,4).NMDA型グルタミン酸受容体はNR1サブユニット,NR2サブユニット(NR2A,NR2B,NR2C,NR2Dの4種類),NR3サブユニット(NR3A,NR3Bの2種類)より構成され,NR2サブユニットの違いがのちの細胞の生死の運命を決定する,すなわち,NR2Aサブユニットの活性化が細胞保護に,NR2Bサブユニットの活性化が細胞障害につながることが多く報告されてきている2,4).
転写因子CREBは,そのSer133がリン酸化されることによってヒストンアセチル化酵素活性をもつコアクチベーターCBP/p300がリクルートされて転写が開始されるため5),このSer133リン酸化を中心とした研究が行われてきた.しかしながら,CREBのSer133リン酸化は転写開始に必要であるが十分ではなく,CBP/p300についてもそのほか多くの転写因子のコアクチベーターとしても作用する.しかしながら近年,CREBに特異的なコアクチベーターであるTORC(別名CRTC,TORC1~TORC3の3つのアイソフォームをもつ)が同定され6,7),CREBの転写制御機構の解明は新たな局面をむかえた.CBP/p300によるCREBの制御はKIDドメイン-KIXドメインを介しているのに対し,TORCファミリーによるCREBの制御はCREBのbZIPドメインを介して行われている.TORCファミリーは刺激のない状態においておもに細胞質にある(TORC3はつねに核に存在する)が,Ca2+あるいはcAMPによる刺激により脱リン酸化が起こり核に移行してCREの転写を活性化する8).
一方で,SIKは高食塩負荷されたラットの副腎から単離されたAMP活性化プロテインキナーゼファミリーに属するキナーゼで9),SIK1~SIK3の3つのアイソフォームをもち,筆者らのグループは,このSIKの基質がTORCファミリーであることを同定してきた8).SIK-TORC経路は糖代謝などにおいてきわめて重要な役割をはたしていることがすでに報告されていたが,中枢神経系においてCREBを介した神経保護あるいは病態にどのような役割をはたしているかはわかっていなかった.そこで,この研究においては,脳虚血をモデルとして中枢神経系におけるSIK-TORC1経路の制御機構および意義を,SIK2ノックアウトマウスを作製しin vitroおよびin vivoにおいて検討した.
in vitroでの脳虚血のモデルである低酸素低グルコース負荷により,CREBのSer133リン酸化は低酸素低グルコース負荷後3~6時間をピークとして上昇し12時間後には対照まで低下したが,一方で,CREの転写活性の上昇は12時間後まで持続していた.低酸素低グルコース負荷後の全長CREB(TORC活性能をもつ)とbZIPドメイン欠損CREB(TORC活性能をもたない)の動態を調べると,bZIPドメイン欠損CREBは全長CREBと比べ活性が有意に低下していた.他方,シクロスポリンAあるいはFK506によるカルシニューリンの抑制はCREBのSer133リン酸化を亢進する傾向をもつのに対しCREの転写活性は有意に抑制され,また,低濃度のスタウロスポリン(10 nM)はCREBのSer133リン酸化を有意に抑制したが,CREの転写活性は著明に亢進した.このことは,大脳皮質のニューロンにおいて,Ser133リン酸化CREBとは独立した,CREBのbZIPドメインを介した転写制御機構の存在することを意味した.
初代大脳皮質ニューロンではTORC1が高発現しており,TORC1は細胞質にあり低酸素低グルコース負荷によりSer167が脱リン酸化され核に移行する.ドミナントネガティブ型TORC1(TORC1のN末端56残基)のアデノウイルスによる導入は低酸素低グルコース負荷後のCREの転写活性を有意に低下させニューロン死を増悪させた.野生型TORC1およびSer167をAlaに置換したTORC1変異体の導入はCREの転写活性を上昇させ,低酸素低グルコース負荷後のニューロンの障害を有意に改善した.また,TORC1の強制発現はCREBに依存的に下流のBDNFおよびPGC-1αの発現を誘導した.
TORCファミリーはSIKファミリーまたはAMP活性化プロテインキナーゼによりリン酸化される8).SIKファミリーのうち,SIK2はmRNAレベルおよびタンパク質レベルにおいて大脳皮質および海馬にて高発現していた.つぎに,低酸素低グルコース負荷後のTORC1の制御に関与するキナーゼを検討した.低酸素低グルコース負荷後の早い時期よりSIK2タンパク質が低下し,これはTORC1のSer167脱リン酸化および活性化と経時変化が一致するため,SIK2がTORC1-CREB経路を介した活性制御において重要であると考えた.まず,ニューロンにおけるSIK2のTORC1制御における重要性を検討するため,低分子キナーゼ阻害剤ライブラリーを用いSIK2をより選択的に阻害する薬物をスクリーニングした.その結果,AMP活性化プロテインキナーゼ阻害剤として知られるCompound Cが低濃度でSIK2を阻害することがわかった(IC50は0.3μM).0.3~0.5μMのCompound Cは低酸素低グルコース負荷後のCREの転写活性を有意に上昇させニューロン保護効果を示した.また,ドミナントネガティブ型SIK2変異体は低酸素低グルコース負荷後のニューロンの障害を増悪させ,キナーゼ活性欠損SIK2変異体はニューロン死を改善した.また,内因性のSIK2をRNAi法により阻害すると低酸素低グルコース負荷後のCREの転写活性の上昇ならびにニューロン保護効果を認めた.
さらにTORC1による制御機構を検討するため,各種のキナーゼ阻害剤を用いてTORC1の活性を検討した.その結果,Ca2+/カルモジュリン依存性キナーゼIIおよびCa2+/カルモジュリン依存性キナーゼIVの阻害剤であるKN93がTORC1の活性を有意に抑制し,また,Ca2+/カルモジュリン依存性キナーゼのなかではドミナントアクティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントアクティブ型Ca2+/カルモジュリン依存性キナーゼIVがTORC1の活性を有意に上昇させた.一方で,ドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIVは低酸素低グルコース負荷後のCREの転写活性を有意に抑制した.また,内因性のCa2+/カルモジュリン依存性キナーゼIVのRNAi法による阻害はこれらの実験結果を裏づけた.このことより,ニューロンにおいてはCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVがSIK2の負の制御キナーゼであることが示された.
ニューロンへのCa2+の流入経路としてNMDA型グルタミン酸受容体の重要性は以前より指摘されていたが,NMDA型グルタミン酸受容体のアンタゴニストであるAP5,NR2AサブユニットをもつNMDA型グルタミン酸受容体のアンタゴニストであるNVP-AM0077,NR2BサブユニットをもつNMDA型グルタミン酸受容体のアンタゴニストであるRo25-6981,非NMDA型グルタミン酸受容体のアンタゴニストであるCNQX,L型Ca2+チャネル遮断剤であるニフェジピンなどを用いて検討したところ,AP5およびNVP-AM0077により有意に低酸素低グルコース負荷後のTORC1活性を抑制した.一方で,ビククリンと4-アミノピリジンによりNR2AサブユニットをもつNMDA型グルタミン酸受容体を刺激したところ2),SIK2の分解およびTORC1の活性化をひき起こした.また,ドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIVはビククリンと4-アミノピリジンによるTORC1の活性化を阻害した.
低酸素低グルコース負荷後のSIK2タンパク質の低下はプロテアソーム阻害剤であるラクタシスチンにより抑制されるためユビキチン-プロテアソーム系を介したSIK2の分解であることが示された10).Ca2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVはSIK2タンパク質を減少させるが,新たにCa2+/カルモジュリン依存性キナーゼモチーフとしてSIK2のThr484がCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVによりリン酸化されてSIK2を負に制御することが示された.このThr484は線虫から哺乳類にいたるまで保存されており非常に重要な部位であることが示唆された.
これまでに述べた結果より,SIK2はCREB-TORC1経路を介して神経保護効果を制御していることが示されたが,さらにin vivoにおけるSIK2の重要性を調べるためSIK2ノックアウトマウスを作製した.まず,SIK2ノックアウトマウスおよび野生型マウスより初代大脳皮質ニューロン培養を行い検討した.SIK2ノックアウトマウスは野生型マウスに比べて,低酸素低グルコース負荷後のCREの転写活性の上昇,TORC1の活性化の上昇,ならびに,低酸素低グルコース負荷後のニューロン死の軽減,下流のPGC-1α,BDNF,TrkBなどの発現の有意な上昇を認めた.
SIK2のin vivoにおける神経保護効果への影響を検討するため,SIK2ノックアウトマウスおよび野生型マウスにおいて60分間の一過性中大脳動脈閉塞モデルを作製した.SIK2ノックアウトマウスにおいては虚血ののちのTORC1のSer167リン酸化レベルが野生型マウスに比べて有意に減少し,梗塞サイズも有意に小さかった.また,SIK2ノックアウトマウスは野生型マウスに比べて大脳皮質ペナンブナ領域におけるPGC-1αおよびBDNFの発現の有意な上昇,ならびに,TNFαの発現の有意な低下を認めた.
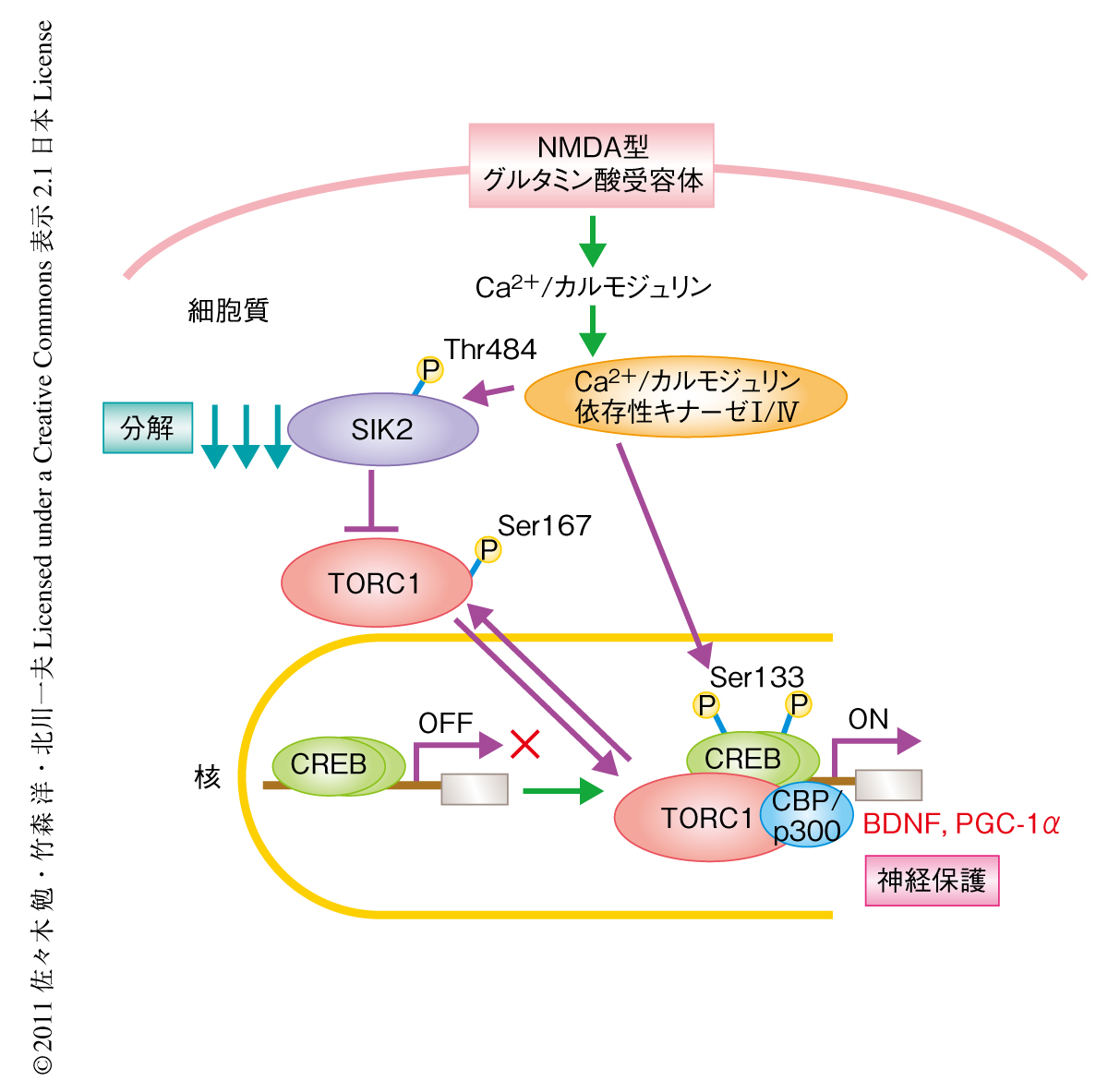
ニューロンにおいて従来まで報告されていたCREBのSer133リン酸化の重要性にくわえて,SIK2によるTORC1-CREB経路の制御の重要性も示された(図1).今回,筆者らが明らかにしたCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIV→SIK2の分解→TORC1の活性化という経路は,脳虚血の内因性保護シグナル,あるいは,シナプスのNMDA型グルタミン酸受容体(おもにNR2AサブユニットをもつNMDA型グルタミン酸受容体)を介した神経保護シグナルに関与していた(図2).とくに,Ca2+/カルモジュリン依存性キナーゼIVはCREBのSer133リン酸化の促進,TORC1の活性化,CBPのSer301リン酸化などを介して11),CREBを介した遺伝子発現を制御する.

今回の研究により,Ca2+はSIK2のThr484リン酸化を介し,一方で,cAMPはSIK2のSer587リン酸化を介し,SIK2を負に制御しているが10),その両方の制御がCREBのSer133リン酸化をともなうことは,転写の開始に必要であることにくわえて,CREBの活性化の指標としてSer133リン酸化が検討されてきた理由ともいえるかもしれない.
SIK2はCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVにより分解されCREBの転写をおもに遅い時期になってから制御するが,SCOPはカルパインにより分解されCREBの転写を制御することが報告されている12).ICERを含めた多様なCREB抑制機構の存在は中枢神経系におけるCREBの活性化の重要性を示唆している.今後,SIK2およびTORC1による制御機構をさらに明らかにし,また,それらを標的とした創薬の開発は記憶,脳虚血あるいは認知症を含めた幅広い神経変性疾患に対して有用である可能性をひめている.
略歴:2003年 大阪大学大学院医学系研究科博士課程 修了,2005年 日本学術振興会 特別研究員を経て,2008年より大阪大学大学院医学系研究科 助手(現 助教).
研究テーマ:脳循環代謝,脳卒中,CREBの制御機構の解明と神経疾患の治療への応用.
竹森 洋(Hiroshi Takemori)
医薬基盤研究所創薬基盤研究部 プロジェクトリーダー.
北川 一夫(Kazuo Kitagawa)
大阪大学大学院医学系研究科 准教授.
© 2011 佐々木 勉・竹森 洋・北川一夫 Licensed under CC 表示 2.1 日本
(1大阪大学大学院医学系研究科 神経内科学,2医薬基盤研究所創薬基盤研究部 代謝疾患関連タンパク探索プロジェクト)
email:佐々木 勉,北川一夫
DOI: 10.7875/first.author.2011.029
SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB.
Tsutomu Sasaki, Hiroshi Takemori, Yoshiki Yagita, Yasukazu Terasaki, Tatsuya Uebi, Nanao Horike, Hiroaki Takagi, Teruo Susumu, Hiroshi Teraoka, Ken-ichi Kusano, Osamu Hatano, Naoki Oyama, Yukio Sugiyama, Saburo Sakoda, Kazuo Kitagawa
Neuron, 69, 106-119 (2011)
要 約
転写因子CREBは脳に豊富に発現し,記憶,神経可塑性,細胞の生存など,幅広い神経機能あるいは病態に関与している.また,ニューロンにはSIK2およびTORC1が豊富に発現しており,SIK2はCREBを介した遺伝子発現を抑制している.筆者らは,大脳皮質のニューロンにおいて脳虚血の際にはCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVの活性化によりSIK2が分解され,TORC1-CREB経路を介して下流の神経保護因子の発現が誘導される,という新規のシグナル伝達経路を同定した.また,脳虚血の際のニューロンにおいてはNMDA型グルタミン酸受容体を介したCa2+の流入が重要であるが,とりわけNR2Aサブユニットをもつタイプがシナプス刺激における内因性の保護シグナルに関与していることを示した.また,SIK2ノックアウトマウスにおいてニューロンの生存の促進効果,脳梗塞サイズの縮小を認めた.今回の成果は,SIK2およびTORC1を標的とした治療が,脳梗塞のみならず認知症などを含めた神経変性疾患に対しても有効な治療につながりうる可能性を示唆した.
はじめに
脳虚血は低酸素,酸化ストレス,炎症,グルタミン酸毒性などの複合的なストレスであり,これまでの研究により脳虚血の病態にかかわる多くの因子および経路が解明され多くの神経保護薬が検討されてきたが,臨床的にはいまだ十分であるとはいえない状況にある.とりわけ,虚血性のニューロン死をひき起こす細胞へのCa2+の流入経路としてNMDA型グルタミン酸受容体は重要なもののひとつであるが,MK-801をはじめとした汎グルタミン酸受容体拮抗薬は臨床的には有効な結果を得られなかった.NMDA型グルタミン酸受容体の活性化により下流の神経保護あるいは神経障害につながりうる多くの転写因子の活性がダイナミックに変化する1).とりわけ転写因子CREB(cAMP responsive element-binding protein)の活性化は,筆者らあるいはほかのグループも,重要な神経保護シグナルとして精力的に研究を行ってきた2,3).しかしながら,近年,グルタミン酸受容体のシナプスあるいはシナプス外での局在の差異あるいはサブタイプの違いなどの詳細な検討がなされ,それらの違いにより下流のシグナル伝達あるいは細胞の生死に決定的な違いのあることが報告された2,4).NMDA型グルタミン酸受容体はNR1サブユニット,NR2サブユニット(NR2A,NR2B,NR2C,NR2Dの4種類),NR3サブユニット(NR3A,NR3Bの2種類)より構成され,NR2サブユニットの違いがのちの細胞の生死の運命を決定する,すなわち,NR2Aサブユニットの活性化が細胞保護に,NR2Bサブユニットの活性化が細胞障害につながることが多く報告されてきている2,4).
転写因子CREBは,そのSer133がリン酸化されることによってヒストンアセチル化酵素活性をもつコアクチベーターCBP/p300がリクルートされて転写が開始されるため5),このSer133リン酸化を中心とした研究が行われてきた.しかしながら,CREBのSer133リン酸化は転写開始に必要であるが十分ではなく,CBP/p300についてもそのほか多くの転写因子のコアクチベーターとしても作用する.しかしながら近年,CREBに特異的なコアクチベーターであるTORC(別名CRTC,TORC1~TORC3の3つのアイソフォームをもつ)が同定され6,7),CREBの転写制御機構の解明は新たな局面をむかえた.CBP/p300によるCREBの制御はKIDドメイン-KIXドメインを介しているのに対し,TORCファミリーによるCREBの制御はCREBのbZIPドメインを介して行われている.TORCファミリーは刺激のない状態においておもに細胞質にある(TORC3はつねに核に存在する)が,Ca2+あるいはcAMPによる刺激により脱リン酸化が起こり核に移行してCREの転写を活性化する8).
一方で,SIKは高食塩負荷されたラットの副腎から単離されたAMP活性化プロテインキナーゼファミリーに属するキナーゼで9),SIK1~SIK3の3つのアイソフォームをもち,筆者らのグループは,このSIKの基質がTORCファミリーであることを同定してきた8).SIK-TORC経路は糖代謝などにおいてきわめて重要な役割をはたしていることがすでに報告されていたが,中枢神経系においてCREBを介した神経保護あるいは病態にどのような役割をはたしているかはわかっていなかった.そこで,この研究においては,脳虚血をモデルとして中枢神経系におけるSIK-TORC1経路の制御機構および意義を,SIK2ノックアウトマウスを作製しin vitroおよびin vivoにおいて検討した.
1.脳虚血のin vitroモデルにおけるCREBの動態
in vitroでの脳虚血のモデルである低酸素低グルコース負荷により,CREBのSer133リン酸化は低酸素低グルコース負荷後3~6時間をピークとして上昇し12時間後には対照まで低下したが,一方で,CREの転写活性の上昇は12時間後まで持続していた.低酸素低グルコース負荷後の全長CREB(TORC活性能をもつ)とbZIPドメイン欠損CREB(TORC活性能をもたない)の動態を調べると,bZIPドメイン欠損CREBは全長CREBと比べ活性が有意に低下していた.他方,シクロスポリンAあるいはFK506によるカルシニューリンの抑制はCREBのSer133リン酸化を亢進する傾向をもつのに対しCREの転写活性は有意に抑制され,また,低濃度のスタウロスポリン(10 nM)はCREBのSer133リン酸化を有意に抑制したが,CREの転写活性は著明に亢進した.このことは,大脳皮質のニューロンにおいて,Ser133リン酸化CREBとは独立した,CREBのbZIPドメインを介した転写制御機構の存在することを意味した.
2.低酸素低グルコース負荷によりTORC1のSer167脱リン酸化が起こる
初代大脳皮質ニューロンではTORC1が高発現しており,TORC1は細胞質にあり低酸素低グルコース負荷によりSer167が脱リン酸化され核に移行する.ドミナントネガティブ型TORC1(TORC1のN末端56残基)のアデノウイルスによる導入は低酸素低グルコース負荷後のCREの転写活性を有意に低下させニューロン死を増悪させた.野生型TORC1およびSer167をAlaに置換したTORC1変異体の導入はCREの転写活性を上昇させ,低酸素低グルコース負荷後のニューロンの障害を有意に改善した.また,TORC1の強制発現はCREBに依存的に下流のBDNFおよびPGC-1αの発現を誘導した.
3.SIK2の抑制はニューロン保護効果をもつ
TORCファミリーはSIKファミリーまたはAMP活性化プロテインキナーゼによりリン酸化される8).SIKファミリーのうち,SIK2はmRNAレベルおよびタンパク質レベルにおいて大脳皮質および海馬にて高発現していた.つぎに,低酸素低グルコース負荷後のTORC1の制御に関与するキナーゼを検討した.低酸素低グルコース負荷後の早い時期よりSIK2タンパク質が低下し,これはTORC1のSer167脱リン酸化および活性化と経時変化が一致するため,SIK2がTORC1-CREB経路を介した活性制御において重要であると考えた.まず,ニューロンにおけるSIK2のTORC1制御における重要性を検討するため,低分子キナーゼ阻害剤ライブラリーを用いSIK2をより選択的に阻害する薬物をスクリーニングした.その結果,AMP活性化プロテインキナーゼ阻害剤として知られるCompound Cが低濃度でSIK2を阻害することがわかった(IC50は0.3μM).0.3~0.5μMのCompound Cは低酸素低グルコース負荷後のCREの転写活性を有意に上昇させニューロン保護効果を示した.また,ドミナントネガティブ型SIK2変異体は低酸素低グルコース負荷後のニューロンの障害を増悪させ,キナーゼ活性欠損SIK2変異体はニューロン死を改善した.また,内因性のSIK2をRNAi法により阻害すると低酸素低グルコース負荷後のCREの転写活性の上昇ならびにニューロン保護効果を認めた.
4.大脳皮質のニューロンにおいてCa2+/カルモジュリン依存性キナーゼはTORC1の活性を制御する
さらにTORC1による制御機構を検討するため,各種のキナーゼ阻害剤を用いてTORC1の活性を検討した.その結果,Ca2+/カルモジュリン依存性キナーゼIIおよびCa2+/カルモジュリン依存性キナーゼIVの阻害剤であるKN93がTORC1の活性を有意に抑制し,また,Ca2+/カルモジュリン依存性キナーゼのなかではドミナントアクティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントアクティブ型Ca2+/カルモジュリン依存性キナーゼIVがTORC1の活性を有意に上昇させた.一方で,ドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIVは低酸素低グルコース負荷後のCREの転写活性を有意に抑制した.また,内因性のCa2+/カルモジュリン依存性キナーゼIVのRNAi法による阻害はこれらの実験結果を裏づけた.このことより,ニューロンにおいてはCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVがSIK2の負の制御キナーゼであることが示された.
5.シナプスにおけるNMDA型グルタミン酸受容体の活性化はCa2+/カルモジュリン依存性キナーゼ-SIK2経路を介してTORC1の活性を上昇させる
ニューロンへのCa2+の流入経路としてNMDA型グルタミン酸受容体の重要性は以前より指摘されていたが,NMDA型グルタミン酸受容体のアンタゴニストであるAP5,NR2AサブユニットをもつNMDA型グルタミン酸受容体のアンタゴニストであるNVP-AM0077,NR2BサブユニットをもつNMDA型グルタミン酸受容体のアンタゴニストであるRo25-6981,非NMDA型グルタミン酸受容体のアンタゴニストであるCNQX,L型Ca2+チャネル遮断剤であるニフェジピンなどを用いて検討したところ,AP5およびNVP-AM0077により有意に低酸素低グルコース負荷後のTORC1活性を抑制した.一方で,ビククリンと4-アミノピリジンによりNR2AサブユニットをもつNMDA型グルタミン酸受容体を刺激したところ2),SIK2の分解およびTORC1の活性化をひき起こした.また,ドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIおよびドミナントネガティブ型Ca2+/カルモジュリン依存性キナーゼIVはビククリンと4-アミノピリジンによるTORC1の活性化を阻害した.
6.Ca2+/カルモジュリン依存性キナーゼはSIK2をリン酸化し分解を促進する
低酸素低グルコース負荷後のSIK2タンパク質の低下はプロテアソーム阻害剤であるラクタシスチンにより抑制されるためユビキチン-プロテアソーム系を介したSIK2の分解であることが示された10).Ca2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVはSIK2タンパク質を減少させるが,新たにCa2+/カルモジュリン依存性キナーゼモチーフとしてSIK2のThr484がCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVによりリン酸化されてSIK2を負に制御することが示された.このThr484は線虫から哺乳類にいたるまで保存されており非常に重要な部位であることが示唆された.
7.SIK2ノックアウトマウスにおけるニューロン保護効果
これまでに述べた結果より,SIK2はCREB-TORC1経路を介して神経保護効果を制御していることが示されたが,さらにin vivoにおけるSIK2の重要性を調べるためSIK2ノックアウトマウスを作製した.まず,SIK2ノックアウトマウスおよび野生型マウスより初代大脳皮質ニューロン培養を行い検討した.SIK2ノックアウトマウスは野生型マウスに比べて,低酸素低グルコース負荷後のCREの転写活性の上昇,TORC1の活性化の上昇,ならびに,低酸素低グルコース負荷後のニューロン死の軽減,下流のPGC-1α,BDNF,TrkBなどの発現の有意な上昇を認めた.
8.SIK2ノックアウトマウスにおける中大脳動脈閉塞モデルでは梗塞サイズが縮小する
SIK2のin vivoにおける神経保護効果への影響を検討するため,SIK2ノックアウトマウスおよび野生型マウスにおいて60分間の一過性中大脳動脈閉塞モデルを作製した.SIK2ノックアウトマウスにおいては虚血ののちのTORC1のSer167リン酸化レベルが野生型マウスに比べて有意に減少し,梗塞サイズも有意に小さかった.また,SIK2ノックアウトマウスは野生型マウスに比べて大脳皮質ペナンブナ領域におけるPGC-1αおよびBDNFの発現の有意な上昇,ならびに,TNFαの発現の有意な低下を認めた.
おわりに
ニューロンにおいて従来まで報告されていたCREBのSer133リン酸化の重要性にくわえて,SIK2によるTORC1-CREB経路の制御の重要性も示された(図1).今回,筆者らが明らかにしたCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIV→SIK2の分解→TORC1の活性化という経路は,脳虚血の内因性保護シグナル,あるいは,シナプスのNMDA型グルタミン酸受容体(おもにNR2AサブユニットをもつNMDA型グルタミン酸受容体)を介した神経保護シグナルに関与していた(図2).とくに,Ca2+/カルモジュリン依存性キナーゼIVはCREBのSer133リン酸化の促進,TORC1の活性化,CBPのSer301リン酸化などを介して11),CREBを介した遺伝子発現を制御する.
今回の研究により,Ca2+はSIK2のThr484リン酸化を介し,一方で,cAMPはSIK2のSer587リン酸化を介し,SIK2を負に制御しているが10),その両方の制御がCREBのSer133リン酸化をともなうことは,転写の開始に必要であることにくわえて,CREBの活性化の指標としてSer133リン酸化が検討されてきた理由ともいえるかもしれない.
SIK2はCa2+/カルモジュリン依存性キナーゼIおよびCa2+/カルモジュリン依存性キナーゼIVにより分解されCREBの転写をおもに遅い時期になってから制御するが,SCOPはカルパインにより分解されCREBの転写を制御することが報告されている12).ICERを含めた多様なCREB抑制機構の存在は中枢神経系におけるCREBの活性化の重要性を示唆している.今後,SIK2およびTORC1による制御機構をさらに明らかにし,また,それらを標的とした創薬の開発は記憶,脳虚血あるいは認知症を含めた幅広い神経変性疾患に対して有用である可能性をひめている.
文 献
- Taghibiglou, C., Martin, H. G., Lai, T. W. et al.: Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nat. Med., 15, 1399-1406 (2009)[PubMed]
- Hardingham, G. E., Fukunaga, Y. & Bading, H.: Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci., 5, 405-414 (2002)[PubMed]
- Mabuchi, T., Kitagawa, K., Kuwabara, K. et al.: Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J. Neurosci., 21, 9204-9213 (2001)[PubMed]
- Sattler, R., Xiong, Z., MacDonald, J. F. et al.: Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J. Neurosci., 20, 22-33 (2000)[PubMed]
- Gonzalez, G. A. & Montminy, M. R.: Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell, 59, 675-680 (1989)[PubMed]
- Conkright, M. D., Canettieri, G., Screaton, R. et al.: TORCs: transducers of regulated CREB activity. Mol. Cell, 12, 413-423 (2003)[PubMed]
- Iourgenko, V., Zhang, W., Mickanin, C. et al.: Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc. Natl. Acad. Sci. USA, 100, 12147-12152 (2003)[PubMed]
- Screaton, R. A., Conkright, M. D., Katoh, Y. et al.: The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell, 119, 61-74 (2004)[PubMed]
- Wang, Z., Takemori, H., Halder, S. K. et al.: Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett., 453, 135-139 (1999)[PubMed]
- Katoh Y, Takemori, H., Lin, X. Z. et al.: Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J., 273, 2730-2748 (2006)[PubMed]
- Impey, S., Fong, A. L., Wang, Y. et al.: Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron, 34, 235-244 (2002)[PubMed]
- Shimizu, K., Phan, T., Mansuy, I. M. et al.: Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell, 128, 1219-1229 (2007)[PubMed]
著者プロフィール
略歴:2003年 大阪大学大学院医学系研究科博士課程 修了,2005年 日本学術振興会 特別研究員を経て,2008年より大阪大学大学院医学系研究科 助手(現 助教).
研究テーマ:脳循環代謝,脳卒中,CREBの制御機構の解明と神経疾患の治療への応用.
竹森 洋(Hiroshi Takemori)
医薬基盤研究所創薬基盤研究部 プロジェクトリーダー.
北川 一夫(Kazuo Kitagawa)
大阪大学大学院医学系研究科 准教授.
© 2011 佐々木 勉・竹森 洋・北川一夫 Licensed under CC 表示 2.1 日本
