筋萎縮性側索硬化症の発症にかかわるアネキシンA11の変異
柴田秀樹・牧 正敏
(名古屋大学大学院生命農学研究科 応用分子生命科学専攻分子細胞制御学研究分野)
email:柴田秀樹
DOI: 10.7875/first.author.2017.049
Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis.
Bradley N. Smith, Simon D. Topp, Claudia Fallini, Hideki Shibata, Han-Jou Chen, Claire Troakes, Andrew King, Nicola Ticozzi, Kevin P. Kenna, Athina Soragia-Gkazi, Jack W. Miller, Akane Sato, Diana Marques Dias, Maryangel Jeon, Caroline Vance, Chun Hao Wong, Martina de Majo, Wejdan Kattuah, Jacqueline C. Mitchell, Emma L. Scotter, Nicholas W. Parkin, Peter C. Sapp, Matthew Nolan, Peter J. Nestor, Michael Simpson, Michael Weale, Monkel Lek, Frank Baas, J. M. Vianney de Jong, Anneloor L. M. A. ten Asbroek, Alberto Garcia Redondo, Jesús Esteban-Pérez, Cinzia Tiloca, Federico Verde, Stefano Duga, Nigel Leigh, Hardev Pall, Karen E. Morrison, Ammar Al-Chalabi, Pamela J. Shaw, Janine Kirby, Martin R. Turner, Kevin Talbot, Orla Hardiman, Jonathan D. Glass, Jacqueline De Belleroche, Masatoshi Maki, Stephen E. Moss, Christopher Miller, Cinzia Gellera, Antonia Ratti, Safa Al-Sarraj, Robert H. Brown Jr., Vincenzo Silani, John E. Landers, Christopher E. Shaw
Science Translational Medicine, 9, eaad9157 (2017)
筋委縮性側索硬化症は脳および脊髄における運動ニューロンの選択的な変性および消失により運動麻痺が進行する原因不明の難病である.この研究において,家族性および孤発性の筋委縮性側索硬化症の患者におけるエキソーム解析により原因遺伝子としてアネキシンA11をコードするANXA11遺伝子が同定され,6つのバリアントが見い出された.また,Asp40がGlyに置換したバリアントは共通の創始者変異をもつことが明らかにされた.Asp40がGlyに置換したバリアントをもつ患者において,脊髄の運動ニューロンや海馬の神経軸索などに抗アネキシンA11抗体に陽性の多くの凝集体がみつかった.また,生化学的な解析により,Asp40のGlyへの置換を含む,筋委縮性側索硬化症において同定されたアネキシンA11のN末端領域の変異により,カルサイクリンとの結合活性が低下あるいは亢進することが示された.さらに,Arg235がGlnに置換したアネキシンA11変異体は細胞において不溶性の凝集体を形成した.アネキシンA11は小胞輸送経路において機能することから,アネキシンA11の変異による小胞輸送の制御機構の破綻が筋委縮性側索硬化症の発症の一因である可能性が考えられた.
筋委縮性側索硬化症は生涯における罹患のリスクが400人に1人という神経変性疾患であり,脳および脊髄の運動ニューロンが選択的かつ進行性に変性および消失することにより運動麻痺を起こし死にいたる難病である.症例の10%は家族性で,欧州の家族性の家系の約60%は原因遺伝子がつきとめられている1,2).孤発性の症例の約10%においては家族性の家系において同定された原因遺伝子に変異をもち,このデータは不完全浸透度を反映したものと考えられる.家族性と孤発性をあわせた症例の約20%はSOD1遺伝子,TARDBP遺伝子,FUS遺伝子のミスセンス変異,あるいは,C9orf72遺伝子のイントロン領域の6塩基の伸長のいずれかが原因であり,1~3%の症例においてはそのほかの原因遺伝子が同定されている3).最近の全ゲノム解析およびエキソーム解析による筋委縮性側索硬化症の家系の連鎖解析によりVCP遺伝子,PFN1遺伝子,MATR3遺伝子,CHCHD10遺伝子,CCNF遺伝子との関連が,また,まれなバリアントを解析することによりTUBA4A遺伝子,TBK1遺伝子,NEK1遺伝子,C21orf2遺伝子との関連が明らかにされている.
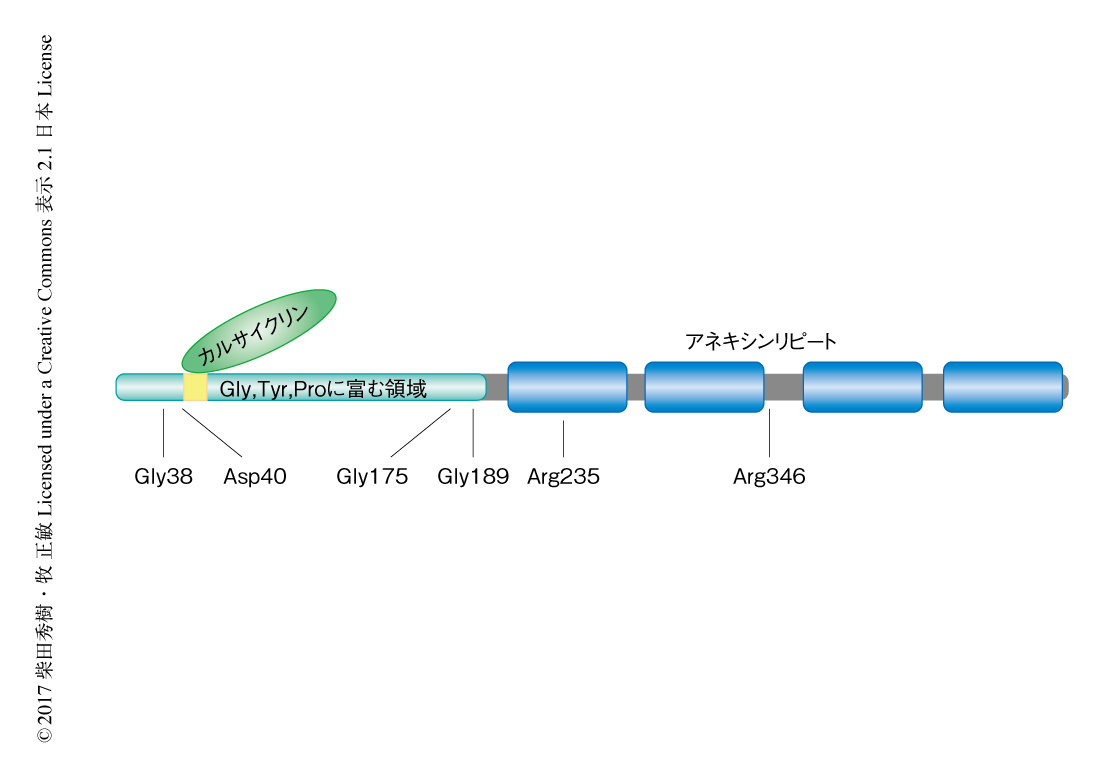
アネキシンA11はヒトが12種もつアネキシンファミリータンパク質のひとつであり,505アミノ酸残基からなり広範な組織に発現する4)(図1).アネキシンファミリータンパク質はC末端側によく保存された4つのアネキシンリピートをもち(アネキシンA6は8つ),Ca2+に依存して酸性リン脂質と結合する.アネキシンA11はアネキシンファミリータンパク質のなかでN末端領域がもっとも長く,Gly,Tyr,Proに富み,カルサイクリン,ALG-2,ソルシンの3つのEFハンドタンパク質がこの領域に結合する.近年,筆者らは,アネキシンA11がALG-2との結合を介して初期分泌経路に動員され小胞輸送を制御することを報告した5,6).この研究においては,筋委縮性側索硬化症の発症にかかわるアネキシンA11変異体がカルサイクリンとの結合に異常をきたすことを見い出した.
筋委縮性側索硬化症の原因遺伝子のひとつであるC9orf72遺伝子における6塩基の伸長をもたない欧州の家族性の筋委縮性側索硬化症のコホートにおけるエキソーム解析から,2人以上が発症している50家系を抽出した.患者において認められ対照には認められないバリアントが1家系あたり平均して10個ほど同定された.これらのなかにはすでに報告されているバリアントが含まれていた.2家系以上で認められたバリアントはわずかに2つであり,ひとつは解析がよく進められているTARDBP遺伝子のバリアント7) であった.もうひとつは新規のアネキシンA11をコードするANXA11遺伝子においてAsp40がGlyに置換したバリアントで,英国の2つの家系において認められた.さらに,のちの解析において,イタリアの1家系においてもこのバリアントが認められた.
類縁関係のない家族性の筋委縮性側索硬化症の患者について3人以上に共有されるバリアントを探索したところ,既知のバリアントとともに,ANXA11遺伝子においてAsp40がGlyに置換したバリアントが3人に認められた.また,英国の孤発性の筋委縮性側索硬化症の患者においてANXA11遺伝子の塩基配列を解析した結果,このバリアントのキャリアが1人同定された.つまり,874人の患者からこのバリアントの4人のキャリアが同定された.シミュレーション解析により,筋委縮性側索硬化症の患者がANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつことは統計的に有意であると推定された.
このバリアントが同定された英国の2家系について,発症した4人を含め17人のエキソームを解析した.その結果,発症していない4人がこのバリアントのキャリアであった.5人のキャリアにおいてANXA11遺伝子の近傍の塩基配列をくわしく解読したところ,4箇所の1塩基多型と2箇所のマイクロサテライト多型からなる共通のハプロタイプの存在が明らかにされた.この4箇所の1塩基多型のハプロタイプから,このバリアントの創始者変異が欧州人のバックグラウンドにおいて生じたことが考えられた.このハプロタイプの領域には23の遺伝子が含まれたが,どのキャリアもほかの遺伝子にはアミノ酸配列に影響を及ぼす変異をもたなかったことから,ANXA11遺伝子におけるAsp40のGlyへの置換が筋委縮性側索硬化症の責任変異と考えられた.
この研究に用いた家族性の筋委縮性側索硬化症のコホートにおいて,血縁関係のない2人の患者にANXA11遺伝子においてGly38がArgに置換したバリアントが認められた.また,Gly175がArgに置換したバリアント,および,Arg346がCysに置換したバリアントが1例ずつ同定された.Gly175がArgに置換したバリアントは同胞の患者において共有されていた.これらのANXA11遺伝子のバリアントをもつ患者は,すでに報告されている17の筋委縮性側索硬化症の原因遺伝子のエキソンには変異が認められなかった.
さらに,英国の孤発性の筋委縮性側索硬化症の患者についてANXA11遺伝子の塩基配列を解析したところ,Asp40がGlyに置換したバリアントとともに,Gly189がGluに置換したバリアント,および,Arg235がGlnに置換したバリアントが1例ずつ確認された.あわせて6つのアネキシンA11の変異が同定されたが,4つはN末端領域にあり,2つはC末端側のアネキシンリピートにあった(図1).変異の生じたアミノ酸残基は塩基配列の解析された哺乳動物において保存されていた.Arg230がCysに置換する1塩基置換は全身性の自己免疫疾患であるサルコイドーシスの発症と強い相関を示すことが報告されているが8),ANXA11遺伝子に変異をもつ筋委縮性側索硬化症の症例においてはみられなかった.
ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ筋委縮性側索硬化症の患者は晩期の発症で認知症状をともなわなかった.このバリアントをもつ6人の症例のうち5人は初期の症状として発語および嚥下機能の低下があった.このバリアントをもつ孤発性の患者の骨髄を組織病理学的に解析したところ,筋委縮性側索硬化症の典型的な所見とともに,抗アネキシンA11抗体に陽性の比較的大きな凝集体が観察された.凝集体の形状は,糸状体様であったり,口径の大きな環形構造であったり,また,繊維状あるいはバスケット状の構造体であったりした.脊髄に比べて数は少ないながら,抗アネキシンA11抗体に陽性の凝集体は運動皮質,海馬歯状回,側頭葉新皮質にも観察され,神経線維網においてはトルペド様の突起構造をともなっていた.さらに,数は少ないが後頭葉においても抗アネキシンA11抗体に陽性の凝集体が観察された.小脳は抗アネキシンA11抗体により染色されなかった.抗アネキシンA11抗体に陽性の凝集体は,ANXA11遺伝子に変異のない筋委縮性側索硬化症の患者,アルツハイマー病の患者,パーキンソン病の患者,対照においては観察されなかった.ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ患者の脊髄前角内はニューロンの脱落が顕著であり,前皮質脊髄路および外側皮質脊髄路に髄鞘の淡明化およびアストログリアの異常な増殖が認められた.残っていた運動ニューロンの多くには抗p62抗体および抗リン酸化TDP-43抗体に陽性の凝集体が観察された.抗リン酸化TDP-43抗体および抗アネキシンA11抗体による二重染色の結果,抗TDP-43抗体に陽性の凝集体と抗アネキシンA11抗体に陽性の凝集体は局在が一致しないことがわかった.一方で,抗アネキシン抗体に陽性の凝集体の一部は抗ユビキチン抗体により染色されたことから,この凝集体のなかにはユビキチン化されたタンパク質の存在する可能性があった.
マウスの初代培養運動ニューロンに,HAタグを付加した野生型のアネキシンA11,および,Gly38がArgに置換したアネキシンA11変異体,Asp40がGlyに置換したアネキシンA11変異体,Arg235がGlnに置換したアネキシンA11変異体をそれぞれ発現させ,抗HAタグ抗体により免疫染色した.その結果,野生型アネキシンA11は細胞体の核と細胞質,軸索,樹状突起に一様に分散し,細胞質においては細胞あたり数個の大きな小胞状の構造体および小さな斑点状の局在様式を示したのに対し,Arg235がGlnに置換したアネキシンA11変異体は小胞状の構造を形成せず,小さな斑点が顕著に増加した.また,細胞質の一様な染色像は得られず,それらの斑点状の構造体に凝集していると考えられた.Asp40がGlyに置換したアネキシンA11変異体は野生型のアネキシンA11と同様の局在様式を示したが,Gly38がArgに置換したアネキシンA11変異体は小胞状の構造体の数が有意に減少していた.ヒト胎児腎細胞HEK293細胞に蛍光タンパク質SGFP2を融合させたArg235がGlnに置換したアネキシンA11変異体を発現させると,ニューロンと同様に,細胞質に斑点状の局在を示したが,それらは抗ユビキチン抗体および抗p62抗体に陽性であった.生化学的には,HEK293細胞に発現させたArg235がGlnに置換したアネキシンA11は界面活性剤Nonidet P-40に不溶性の画分に回収され,SDS-ポリアクリルアミドゲル電気泳動により展開すると移動度の遅いラダー状のタンパク質として検出された.
野生型のアネキシンA11がArg235がGlnに置換したアネキシンA11変異体の形成する斑点状の構造体に動員される可能性について検証するため,それらをヒト神経芽細胞腫SH-SY5Y細胞に共発現させて局在を観察した.その結果,Arg235がGlnに置換したアネキシンA11変異体の斑点状の構造体に野生型のアネキシンA11の一部が動員されている染色像が得られた.また,HEK293細胞に同様に共発現させると,野生型のアネキシンA11はArg235がGlnに置換したアネキシンA11変異体とともに不溶性の画分に回収された.これらの結果から,Arg235がGlnに置換したアネキシンA11変異体は野生型のアネキシンA11を細胞質の不溶性凝集体に動員することにより,ドミナントネガティブ変異体として細胞毒性を生じているのかもしれない.
アネキシンA11のN末端側の50~62残基の領域にはEFハンドを2つもつCa2+結合タンパク質であるカルサイクリンが結合する8)(図1).この領域に近いGly38およびAsp40における変異はアネキシンA11とカルサイクリンとの結合に影響を及ぼす可能性があった.このことを検証するため,野生型のアネキシンA11あるいはアネキシンA11変異体とFLAGタグを付加したカルサイクリンとをHEK293細胞に発現させ,Ca2+の存在下において抗FLAG抗体により免疫沈降しアネキシンA11の結合量を比較した.その結果,野生型のアネキシンA11およびGly38がArgに置換したアネキシンA11変異体はカルサイクリンと共沈降したが,その量は野生型のアネキシンA11よりGly38がArgに置換したアネキシンA11変異体のほうが多かった.一方,Asp40がGlyに置換したアネキシンA11変異体,Gly189がGluに置換したアネキシンA11変異体,Arg235がGlnに置換したアネキシンA11変異体はカルサイクリンとほとんど共沈降しなかった.また,筋委縮性側索硬化症と関連のないアネキシンA11変異体のカルサイクリンとの結合量は野生型のアネキシンA11と同等であった.さらに,アネキシンA11と結合するALG-2およびソルシンについても同様に検討したが,これらとの結合についてはアネキシンA11の変異による影響はみられなかった.
カルサイクリンとの結合がアネキシンA11の可溶性におよぼす影響について解析するため,HEK293細胞に野生型のアネキシンA11あるいはArg235がGlnに置換したアネキシンA11変異体とカルサイクリンとを発現させ,細胞を溶解してこれらのタンパク質の可溶性について検討した.その結果,カルサイクリンとの共発現により,野生型のアネキシンA11およびArg235がGlnに置換したアネキシンA11変異体の発現量は低下し,また,Arg235がGlnに置換したアネキシンA11変異体はすべて可溶性の画分に回収された.プロテアソームの阻害剤であるMG132を添加することによりアネキシンA11の発現量は回復し,Arg235がGlnに置換したアネキシンA11変異体は不溶性の画分に検出されたことから,カルサイクリンはユビキチン-プロテアソーム系を介してアネキシンA11の発現量を制御している可能性が考えられた.
ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ筋委縮性側索硬化症の患者の脊髄を抗カルサイクリン抗体により染色したところ,ニューロンの染色像は対照と同様であったが,外側皮質脊髄路のアストロサイトの細胞質におけるシグナルが顕著に増強していた.皮質脊髄路のアストロサイトにおけるカルサイクリンの発現量の増加は,孤発性の筋委縮性側索硬化症の症例や,SOD1遺伝子においてGly93がAlaに置換したトランスジェニックマウスにおいても報告されているが9,10),このアストロサイトにおけるカルサイクリンの発現量の増加の機能的な意義については不明である.
この研究において,家族性および孤発性の筋委縮性側索硬化症の患者からその原因となるANXA11遺伝子におけるまれなバリアントを6つ同定した(図1).この研究に用いたコホートにおいては,ANXA11遺伝子の変異が家族性の患者の約1%,孤発性の患者の1.7%に認められたことになる.このうち4つの変異体は,カルサイクリンとの結合活性が野生型よりも低下あるいは亢進した.ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ患者においてはこれまでに報告のない抗アネキシンA11抗体に陽性の凝集体が多く観察され,また,それらとは別に,抗リン酸化TDP-43抗体に陽性の凝集体も検出された.アネキシンA11の細胞における動態はカルサイクリンとの結合により制御されるのかもしれない.今後,同定された筋委縮性側索硬化症の原因となるアネキシンA11の変異が,アネキシンA11のフォールディング,発現量,小胞輸送経路における機能にどのような影響をおよぼすのか,また,なぜTDP-43の凝集が起こるのかなど,筋委縮性側索硬化症の発症の分子機構の解明につながる詳細な解析が待たれる.
略歴:2001年 神戸大学大学院自然科学研究科にて博士号取得,神戸大学理学部 助手,名古屋大学大学院生命農学研究科 助手を経て,2010年より同 准教授.
研究テーマ:Ca2+シグナル伝達とメンブレントラフィック.
関心事:さまざまな環境におかれる細胞の恒常性の維持の機構およびその破綻のもたらす生体への影響.
牧 正敏(Masatoshi Maki)
名古屋大学大学院生命農学研究科 教授.
© 2017 柴田秀樹・牧 正敏 Licensed under CC 表示 2.1 日本
(名古屋大学大学院生命農学研究科 応用分子生命科学専攻分子細胞制御学研究分野)
email:柴田秀樹
DOI: 10.7875/first.author.2017.049
Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis.
Bradley N. Smith, Simon D. Topp, Claudia Fallini, Hideki Shibata, Han-Jou Chen, Claire Troakes, Andrew King, Nicola Ticozzi, Kevin P. Kenna, Athina Soragia-Gkazi, Jack W. Miller, Akane Sato, Diana Marques Dias, Maryangel Jeon, Caroline Vance, Chun Hao Wong, Martina de Majo, Wejdan Kattuah, Jacqueline C. Mitchell, Emma L. Scotter, Nicholas W. Parkin, Peter C. Sapp, Matthew Nolan, Peter J. Nestor, Michael Simpson, Michael Weale, Monkel Lek, Frank Baas, J. M. Vianney de Jong, Anneloor L. M. A. ten Asbroek, Alberto Garcia Redondo, Jesús Esteban-Pérez, Cinzia Tiloca, Federico Verde, Stefano Duga, Nigel Leigh, Hardev Pall, Karen E. Morrison, Ammar Al-Chalabi, Pamela J. Shaw, Janine Kirby, Martin R. Turner, Kevin Talbot, Orla Hardiman, Jonathan D. Glass, Jacqueline De Belleroche, Masatoshi Maki, Stephen E. Moss, Christopher Miller, Cinzia Gellera, Antonia Ratti, Safa Al-Sarraj, Robert H. Brown Jr., Vincenzo Silani, John E. Landers, Christopher E. Shaw
Science Translational Medicine, 9, eaad9157 (2017)
要 約
筋委縮性側索硬化症は脳および脊髄における運動ニューロンの選択的な変性および消失により運動麻痺が進行する原因不明の難病である.この研究において,家族性および孤発性の筋委縮性側索硬化症の患者におけるエキソーム解析により原因遺伝子としてアネキシンA11をコードするANXA11遺伝子が同定され,6つのバリアントが見い出された.また,Asp40がGlyに置換したバリアントは共通の創始者変異をもつことが明らかにされた.Asp40がGlyに置換したバリアントをもつ患者において,脊髄の運動ニューロンや海馬の神経軸索などに抗アネキシンA11抗体に陽性の多くの凝集体がみつかった.また,生化学的な解析により,Asp40のGlyへの置換を含む,筋委縮性側索硬化症において同定されたアネキシンA11のN末端領域の変異により,カルサイクリンとの結合活性が低下あるいは亢進することが示された.さらに,Arg235がGlnに置換したアネキシンA11変異体は細胞において不溶性の凝集体を形成した.アネキシンA11は小胞輸送経路において機能することから,アネキシンA11の変異による小胞輸送の制御機構の破綻が筋委縮性側索硬化症の発症の一因である可能性が考えられた.
はじめに
筋委縮性側索硬化症は生涯における罹患のリスクが400人に1人という神経変性疾患であり,脳および脊髄の運動ニューロンが選択的かつ進行性に変性および消失することにより運動麻痺を起こし死にいたる難病である.症例の10%は家族性で,欧州の家族性の家系の約60%は原因遺伝子がつきとめられている1,2).孤発性の症例の約10%においては家族性の家系において同定された原因遺伝子に変異をもち,このデータは不完全浸透度を反映したものと考えられる.家族性と孤発性をあわせた症例の約20%はSOD1遺伝子,TARDBP遺伝子,FUS遺伝子のミスセンス変異,あるいは,C9orf72遺伝子のイントロン領域の6塩基の伸長のいずれかが原因であり,1~3%の症例においてはそのほかの原因遺伝子が同定されている3).最近の全ゲノム解析およびエキソーム解析による筋委縮性側索硬化症の家系の連鎖解析によりVCP遺伝子,PFN1遺伝子,MATR3遺伝子,CHCHD10遺伝子,CCNF遺伝子との関連が,また,まれなバリアントを解析することによりTUBA4A遺伝子,TBK1遺伝子,NEK1遺伝子,C21orf2遺伝子との関連が明らかにされている.
アネキシンA11はヒトが12種もつアネキシンファミリータンパク質のひとつであり,505アミノ酸残基からなり広範な組織に発現する4)(図1).アネキシンファミリータンパク質はC末端側によく保存された4つのアネキシンリピートをもち(アネキシンA6は8つ),Ca2+に依存して酸性リン脂質と結合する.アネキシンA11はアネキシンファミリータンパク質のなかでN末端領域がもっとも長く,Gly,Tyr,Proに富み,カルサイクリン,ALG-2,ソルシンの3つのEFハンドタンパク質がこの領域に結合する.近年,筆者らは,アネキシンA11がALG-2との結合を介して初期分泌経路に動員され小胞輸送を制御することを報告した5,6).この研究においては,筋委縮性側索硬化症の発症にかかわるアネキシンA11変異体がカルサイクリンとの結合に異常をきたすことを見い出した.
1.筋委縮性側索硬化症の患者のエキソーム解析によるANXA11遺伝子におけるミスセンス変異の同定
筋委縮性側索硬化症の原因遺伝子のひとつであるC9orf72遺伝子における6塩基の伸長をもたない欧州の家族性の筋委縮性側索硬化症のコホートにおけるエキソーム解析から,2人以上が発症している50家系を抽出した.患者において認められ対照には認められないバリアントが1家系あたり平均して10個ほど同定された.これらのなかにはすでに報告されているバリアントが含まれていた.2家系以上で認められたバリアントはわずかに2つであり,ひとつは解析がよく進められているTARDBP遺伝子のバリアント7) であった.もうひとつは新規のアネキシンA11をコードするANXA11遺伝子においてAsp40がGlyに置換したバリアントで,英国の2つの家系において認められた.さらに,のちの解析において,イタリアの1家系においてもこのバリアントが認められた.
類縁関係のない家族性の筋委縮性側索硬化症の患者について3人以上に共有されるバリアントを探索したところ,既知のバリアントとともに,ANXA11遺伝子においてAsp40がGlyに置換したバリアントが3人に認められた.また,英国の孤発性の筋委縮性側索硬化症の患者においてANXA11遺伝子の塩基配列を解析した結果,このバリアントのキャリアが1人同定された.つまり,874人の患者からこのバリアントの4人のキャリアが同定された.シミュレーション解析により,筋委縮性側索硬化症の患者がANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつことは統計的に有意であると推定された.
このバリアントが同定された英国の2家系について,発症した4人を含め17人のエキソームを解析した.その結果,発症していない4人がこのバリアントのキャリアであった.5人のキャリアにおいてANXA11遺伝子の近傍の塩基配列をくわしく解読したところ,4箇所の1塩基多型と2箇所のマイクロサテライト多型からなる共通のハプロタイプの存在が明らかにされた.この4箇所の1塩基多型のハプロタイプから,このバリアントの創始者変異が欧州人のバックグラウンドにおいて生じたことが考えられた.このハプロタイプの領域には23の遺伝子が含まれたが,どのキャリアもほかの遺伝子にはアミノ酸配列に影響を及ぼす変異をもたなかったことから,ANXA11遺伝子におけるAsp40のGlyへの置換が筋委縮性側索硬化症の責任変異と考えられた.
2.家族性および孤発性の筋委縮性側索硬化症の患者において認められたANXA11遺伝子のバリアント
この研究に用いた家族性の筋委縮性側索硬化症のコホートにおいて,血縁関係のない2人の患者にANXA11遺伝子においてGly38がArgに置換したバリアントが認められた.また,Gly175がArgに置換したバリアント,および,Arg346がCysに置換したバリアントが1例ずつ同定された.Gly175がArgに置換したバリアントは同胞の患者において共有されていた.これらのANXA11遺伝子のバリアントをもつ患者は,すでに報告されている17の筋委縮性側索硬化症の原因遺伝子のエキソンには変異が認められなかった.
さらに,英国の孤発性の筋委縮性側索硬化症の患者についてANXA11遺伝子の塩基配列を解析したところ,Asp40がGlyに置換したバリアントとともに,Gly189がGluに置換したバリアント,および,Arg235がGlnに置換したバリアントが1例ずつ確認された.あわせて6つのアネキシンA11の変異が同定されたが,4つはN末端領域にあり,2つはC末端側のアネキシンリピートにあった(図1).変異の生じたアミノ酸残基は塩基配列の解析された哺乳動物において保存されていた.Arg230がCysに置換する1塩基置換は全身性の自己免疫疾患であるサルコイドーシスの発症と強い相関を示すことが報告されているが8),ANXA11遺伝子に変異をもつ筋委縮性側索硬化症の症例においてはみられなかった.
3.ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ筋委縮性側索硬化症の患者の病態および病理学的な特徴
ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ筋委縮性側索硬化症の患者は晩期の発症で認知症状をともなわなかった.このバリアントをもつ6人の症例のうち5人は初期の症状として発語および嚥下機能の低下があった.このバリアントをもつ孤発性の患者の骨髄を組織病理学的に解析したところ,筋委縮性側索硬化症の典型的な所見とともに,抗アネキシンA11抗体に陽性の比較的大きな凝集体が観察された.凝集体の形状は,糸状体様であったり,口径の大きな環形構造であったり,また,繊維状あるいはバスケット状の構造体であったりした.脊髄に比べて数は少ないながら,抗アネキシンA11抗体に陽性の凝集体は運動皮質,海馬歯状回,側頭葉新皮質にも観察され,神経線維網においてはトルペド様の突起構造をともなっていた.さらに,数は少ないが後頭葉においても抗アネキシンA11抗体に陽性の凝集体が観察された.小脳は抗アネキシンA11抗体により染色されなかった.抗アネキシンA11抗体に陽性の凝集体は,ANXA11遺伝子に変異のない筋委縮性側索硬化症の患者,アルツハイマー病の患者,パーキンソン病の患者,対照においては観察されなかった.ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ患者の脊髄前角内はニューロンの脱落が顕著であり,前皮質脊髄路および外側皮質脊髄路に髄鞘の淡明化およびアストログリアの異常な増殖が認められた.残っていた運動ニューロンの多くには抗p62抗体および抗リン酸化TDP-43抗体に陽性の凝集体が観察された.抗リン酸化TDP-43抗体および抗アネキシンA11抗体による二重染色の結果,抗TDP-43抗体に陽性の凝集体と抗アネキシンA11抗体に陽性の凝集体は局在が一致しないことがわかった.一方で,抗アネキシン抗体に陽性の凝集体の一部は抗ユビキチン抗体により染色されたことから,この凝集体のなかにはユビキチン化されたタンパク質の存在する可能性があった.
4.アネキシンA11変異体の生化学的および細胞生物学的な特徴
マウスの初代培養運動ニューロンに,HAタグを付加した野生型のアネキシンA11,および,Gly38がArgに置換したアネキシンA11変異体,Asp40がGlyに置換したアネキシンA11変異体,Arg235がGlnに置換したアネキシンA11変異体をそれぞれ発現させ,抗HAタグ抗体により免疫染色した.その結果,野生型アネキシンA11は細胞体の核と細胞質,軸索,樹状突起に一様に分散し,細胞質においては細胞あたり数個の大きな小胞状の構造体および小さな斑点状の局在様式を示したのに対し,Arg235がGlnに置換したアネキシンA11変異体は小胞状の構造を形成せず,小さな斑点が顕著に増加した.また,細胞質の一様な染色像は得られず,それらの斑点状の構造体に凝集していると考えられた.Asp40がGlyに置換したアネキシンA11変異体は野生型のアネキシンA11と同様の局在様式を示したが,Gly38がArgに置換したアネキシンA11変異体は小胞状の構造体の数が有意に減少していた.ヒト胎児腎細胞HEK293細胞に蛍光タンパク質SGFP2を融合させたArg235がGlnに置換したアネキシンA11変異体を発現させると,ニューロンと同様に,細胞質に斑点状の局在を示したが,それらは抗ユビキチン抗体および抗p62抗体に陽性であった.生化学的には,HEK293細胞に発現させたArg235がGlnに置換したアネキシンA11は界面活性剤Nonidet P-40に不溶性の画分に回収され,SDS-ポリアクリルアミドゲル電気泳動により展開すると移動度の遅いラダー状のタンパク質として検出された.
野生型のアネキシンA11がArg235がGlnに置換したアネキシンA11変異体の形成する斑点状の構造体に動員される可能性について検証するため,それらをヒト神経芽細胞腫SH-SY5Y細胞に共発現させて局在を観察した.その結果,Arg235がGlnに置換したアネキシンA11変異体の斑点状の構造体に野生型のアネキシンA11の一部が動員されている染色像が得られた.また,HEK293細胞に同様に共発現させると,野生型のアネキシンA11はArg235がGlnに置換したアネキシンA11変異体とともに不溶性の画分に回収された.これらの結果から,Arg235がGlnに置換したアネキシンA11変異体は野生型のアネキシンA11を細胞質の不溶性凝集体に動員することにより,ドミナントネガティブ変異体として細胞毒性を生じているのかもしれない.
5.筋委縮性側索硬化症の原因となるアネキシンA11のN末端領域の変異体はカルサイクリンとの結合に異常をきたす
アネキシンA11のN末端側の50~62残基の領域にはEFハンドを2つもつCa2+結合タンパク質であるカルサイクリンが結合する8)(図1).この領域に近いGly38およびAsp40における変異はアネキシンA11とカルサイクリンとの結合に影響を及ぼす可能性があった.このことを検証するため,野生型のアネキシンA11あるいはアネキシンA11変異体とFLAGタグを付加したカルサイクリンとをHEK293細胞に発現させ,Ca2+の存在下において抗FLAG抗体により免疫沈降しアネキシンA11の結合量を比較した.その結果,野生型のアネキシンA11およびGly38がArgに置換したアネキシンA11変異体はカルサイクリンと共沈降したが,その量は野生型のアネキシンA11よりGly38がArgに置換したアネキシンA11変異体のほうが多かった.一方,Asp40がGlyに置換したアネキシンA11変異体,Gly189がGluに置換したアネキシンA11変異体,Arg235がGlnに置換したアネキシンA11変異体はカルサイクリンとほとんど共沈降しなかった.また,筋委縮性側索硬化症と関連のないアネキシンA11変異体のカルサイクリンとの結合量は野生型のアネキシンA11と同等であった.さらに,アネキシンA11と結合するALG-2およびソルシンについても同様に検討したが,これらとの結合についてはアネキシンA11の変異による影響はみられなかった.
カルサイクリンとの結合がアネキシンA11の可溶性におよぼす影響について解析するため,HEK293細胞に野生型のアネキシンA11あるいはArg235がGlnに置換したアネキシンA11変異体とカルサイクリンとを発現させ,細胞を溶解してこれらのタンパク質の可溶性について検討した.その結果,カルサイクリンとの共発現により,野生型のアネキシンA11およびArg235がGlnに置換したアネキシンA11変異体の発現量は低下し,また,Arg235がGlnに置換したアネキシンA11変異体はすべて可溶性の画分に回収された.プロテアソームの阻害剤であるMG132を添加することによりアネキシンA11の発現量は回復し,Arg235がGlnに置換したアネキシンA11変異体は不溶性の画分に検出されたことから,カルサイクリンはユビキチン-プロテアソーム系を介してアネキシンA11の発現量を制御している可能性が考えられた.
ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ筋委縮性側索硬化症の患者の脊髄を抗カルサイクリン抗体により染色したところ,ニューロンの染色像は対照と同様であったが,外側皮質脊髄路のアストロサイトの細胞質におけるシグナルが顕著に増強していた.皮質脊髄路のアストロサイトにおけるカルサイクリンの発現量の増加は,孤発性の筋委縮性側索硬化症の症例や,SOD1遺伝子においてGly93がAlaに置換したトランスジェニックマウスにおいても報告されているが9,10),このアストロサイトにおけるカルサイクリンの発現量の増加の機能的な意義については不明である.
おわりに
この研究において,家族性および孤発性の筋委縮性側索硬化症の患者からその原因となるANXA11遺伝子におけるまれなバリアントを6つ同定した(図1).この研究に用いたコホートにおいては,ANXA11遺伝子の変異が家族性の患者の約1%,孤発性の患者の1.7%に認められたことになる.このうち4つの変異体は,カルサイクリンとの結合活性が野生型よりも低下あるいは亢進した.ANXA11遺伝子においてAsp40がGlyに置換したバリアントをもつ患者においてはこれまでに報告のない抗アネキシンA11抗体に陽性の凝集体が多く観察され,また,それらとは別に,抗リン酸化TDP-43抗体に陽性の凝集体も検出された.アネキシンA11の細胞における動態はカルサイクリンとの結合により制御されるのかもしれない.今後,同定された筋委縮性側索硬化症の原因となるアネキシンA11の変異が,アネキシンA11のフォールディング,発現量,小胞輸送経路における機能にどのような影響をおよぼすのか,また,なぜTDP-43の凝集が起こるのかなど,筋委縮性側索硬化症の発症の分子機構の解明につながる詳細な解析が待たれる.
文 献
- Renton, A. E., Chio, A. & Traynor, B. J.: State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci., 17, 17-23 (2014)[PubMed]
- White, M. A. & Sreedharan, J.: Amyotrophic lateral sclerosis: recent genetic highlights. Curr. Opin. Neurol., 29, 557-564 (2016)[PubMed]
- Smith, B. N., Newhouse, S., Shatunov, A. et al.: The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet., 21, 102-108 (2013)[PubMed]
- Gerke, V., Creutz, C. E. & Moss, S. E.: Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol., 6, 449-461 (2005)[PubMed]
- Shibata, H., Kanadome, T., Sugiura, H. et al.: A new role for annexin A11 in the early secretory pathway via stabilizing Sec31A protein at the endoplasmic reticulum exit sites (ERES). J. Biol. Chem., 290, 4981-4993 (2015)[PubMed]
- Maki, M., Takahara, T. & Shibata, H.: Multifaceted roles of ALG-2 in Ca2+-regulated membrane trafficking. Int. J. Mol. Sci., 17, 1401 (2016)[PubMed]
- Sreedharan, J., Blair, I. P., Tripathi, V. B. et al.: TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 319, 1668-1672 (2008)[PubMed]
- Sudo, T. & Hidaka, H.: Characterization of the calcyclin (S100A6) binding site of annexin XI-A by site-directed mutagenesis. FEBS Lett., 444, 11-14 (1999)[PubMed]
- Hoyaux, D., Boom, A., Van den Bosch, L. et al.: S100A6 overexpression within astrocytes associated with impaired axons from both ALS mouse model and human patients. J. Neuropathol. Exp. Neurol., 61, 736-744 (2002)[PubMed]
- Hoyaux, D., Alao, J., Fuchs, J. et al.: S100A6, a calcium- and zinc-binding protein, is overexpressed in SOD1 mutant mice, a model for amyotrophic lateral sclerosis. Biochim. Biophys. Acta, 1498, 264-272 (2000)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2001年 神戸大学大学院自然科学研究科にて博士号取得,神戸大学理学部 助手,名古屋大学大学院生命農学研究科 助手を経て,2010年より同 准教授.
研究テーマ:Ca2+シグナル伝達とメンブレントラフィック.
関心事:さまざまな環境におかれる細胞の恒常性の維持の機構およびその破綻のもたらす生体への影響.
牧 正敏(Masatoshi Maki)
名古屋大学大学院生命農学研究科 教授.
© 2017 柴田秀樹・牧 正敏 Licensed under CC 表示 2.1 日本
