インスリンシグナルはFoxM1,PLK1,CENP-Aを介して代償性の膵β細胞の増殖を制御する
白川 純・Rohit N. Kulkarni
(米国Harvard大学Joslin Diabetes Center,Section on Islet Cell and Regenerative Biology)
email:白川 純
DOI: 10.7875/first.author.2017.027
Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation.
Jun Shirakawa, Megan Fernandez, Tomozumi Takatani, Abdelfattah El Ouaamari, Prapaporn Jungtrakoon, Erin R. Okawa, Wei Zhang, Peng Yi, Alessandro Doria, Rohit N. Kulkarni
Cell Metabolism, 25, 868-882.e5 (2017)
これまで,膵β細胞の増殖の制御についてはおもにG0期からG1期への移行が注目されていたが,この研究においては,M期における染色体の分離に必須のタンパク質であるCENP-Aに着目した.インスリンシグナルは膵β細胞においてCdk1およびCdk2を介して核にて転写因子であるFoxM1を活性化し,CENP-AおよびPLK1の発現を促進した.セントロメア領域へのCENP-Aの結合にはキナーゼであるPLK1が必須であり,CENP-Aをノックダウンした膵β細胞は増殖の低下およびアポトーシスの亢進を示した.膵β細胞に特異的なCENP-Aノックアウトマウスは加齢,妊娠,高脂肪食の負荷,急性インスリン抵抗性モデルにおいて代償性の膵β細胞の増殖が障害された.2型糖尿病の患者に由来する膵島でおいては健常人に由来する膵島と比較して,インスリンシグナルの活性化によるCENP-AおよびPLK1の発現の誘導が低下した.これより,FoxM1,PLK1,CENP-Aを介する経路は代償性の膵β細胞の増殖において必須であり,糖尿病の進行を抑制する治療の標的になりうると考えられた.
2型糖尿病においては膵β細胞の量の減少が発症および進展に関与しており,膵β細胞の増殖に着目した治療法が注目されている1).これまで,動物モデルにおいて膵β細胞の増殖あるいは生存にインスリンシグナルが重要であることが報告されている.また,2型糖尿病の患者に由来する膵島においては健常人に由来する膵島と比較してインスリンシグナルに関連するタンパク質の発現が低下することが報告されている2,3).
膵β細胞はもっとも高度に分化した細胞のひとつであり,成体においてほとんどの膵β細胞は静止期すなわちG0期にある.インスリンなどの増殖因子はG0期からG1期への移行の促進やG1期/S期チェックポイントの制御に関与することが報告されているが,それ以降のG2期/M期チェックポイントに関してその制御機構は不明であった(図1).
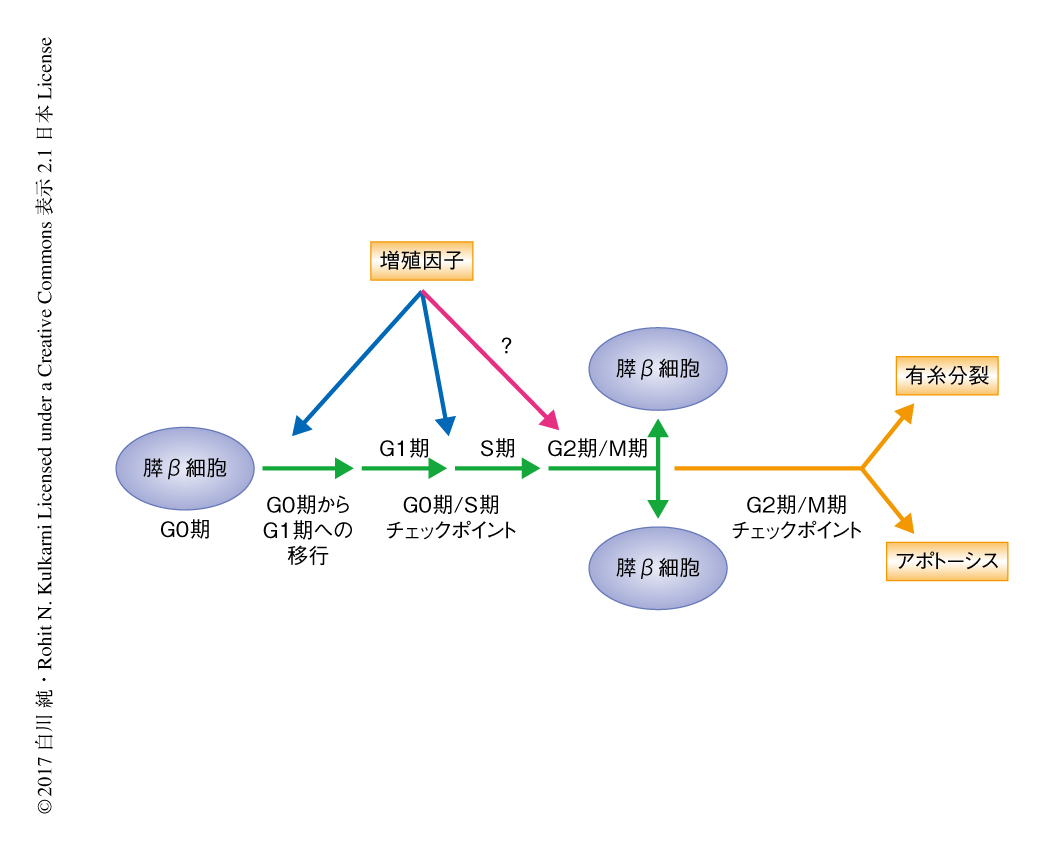
膵β細胞に特異的なインスリン受容体ノックアウトマウスは,膵β細胞におけるインスリンシグナルの低下により高脂肪食の負荷や肝臓に特異的なインスリン抵抗性モデルにおいて代償性の膵β細胞の増殖が障害される2型糖尿病モデルマウスである4).この研究においては,このノックアウトマウスの膵β細胞において発現の低下したCENP-AおよびPLK1に着目した.
インスリン受容体を欠損した膵β細胞において細胞周期を解析したところ,G0期の細胞およびG1期の細胞が増加しておりG0期における細胞周期の停止が示唆された.G2期の細胞とM期の細胞とを分離したところ,インスリン受容体を欠損した膵β細胞においてM期の細胞が有意に増加しておりM期の進行の低下が示唆された.そこで,膵β細胞に特異的なインスリン受容体ノックアウトマウスの膵β細胞においてマイクロアレイ法を用いて遺伝子発現を網羅的に解析したところ,M期における染色体の分離にかかわるCENP-AおよびM期キナーゼのひとつPLK1の発現が野生型の膵β細胞と比べ有意に低下した.
インスリン受容体を欠損した膵β細胞にインスリンとの結合能の低下した変異型のインスリン受容体を発現させたところ,野生型のインスリン受容体を発現させたときと比べ,CENP-AおよびPLK1の発現は低下した.以上より,膵β細胞においてインスリン受容体へのインスリンの結合がCENP-AおよびPLK1の発現を制御することが示された.転写因子であるFoxM1はCENP-AおよびPLK1の発現を制御することが報告されていたが5,6),インスリン受容体を欠損した膵β細胞およびインスリン受容体を欠損した膵島におけるFoxM1の発現は野生型の膵β細胞と同等であった.膵β細胞においてインスリンのほかにもIGF-1あるいはグルコースによる刺激でもCENP-AおよびPLK1の発現は誘導されたが,グルコースによる刺激はインスリン受容体に依存性であった.
FoxM1の細胞における局在およびDNAへの結合能について解析した.マウスにおいてFoxM1は野生型の膵β細胞およびインスリン受容体を欠損した膵β細胞において,インスリンによる刺激の前後とも核に局在しており,MEK1およびMEK2の阻害剤の添加により核の外へと移行した.ヒトの膵β細胞においてFoxM1は刺激をあたえていない状態ではおもに細胞質に局在してしたが,インスリンによる刺激により核への移行が促進され,MEK1およびMEK2の阻害剤の添加により核への移行が阻害された.インスリン受容体を欠損した膵β細胞においてFoxM1の転写活性への関与が報告されていたCdk1およびCdk2の発現は野生型の膵β細胞と比較して有意に低下した.インスリン受容体およびIGF1受容体の阻害剤,PI3キナーゼの阻害剤,Akt1およびAkt2の阻害剤の存在のもとでは,インスリンの刺激によるCdk1およびCdk2,CENP-A,PLK1の発現の誘導は阻害された.
クロマチン免疫沈降法により,FoxM1はCENP-A遺伝子領域およびPLK1遺伝子領域への結合が示されたが,インスリン受容体を欠損した膵β細胞,また,FoxM1の阻害剤あるいはPI3キナーゼの阻害剤の添加によりその結合能は低下した.これより,膵β細胞においてインスリンシグナルは,FoxM1の発現よりもFoxM1の活性を制御することが示唆された.これらインスリンによるCENP-AおよびPLK1の発現の制御は膵β細胞に特異的であり,肝臓あるいは脂肪組織においてはインスリンによる刺激に非依存性であった.
2型糖尿病の患者に由来する膵島においては,健常人に由来する膵島と比較してmRNAレベルでのCENP-Aの発現は有意に低下した.また,免疫染色によりCENP-A陽性の膵β細胞の割合は2型糖尿病の患者に由来する膵島において有意に低下した.健常人に由来する膵島をインスリンにより刺激したところCENP-AおよびPLK1の発現は有意に上昇したが,2型糖尿病の患者に由来する膵島を刺激しても発現は上昇しなかった.
また,家族性の糖尿病を示す52家系に対する全エクソーム解析により,PLK1遺伝子におけるミスセンス変異が原因遺伝子である可能性が示唆された.
MIP-Cre/ERTマウス7) から膵β細胞に特異的なCENP-Aノックアウトマウスを作製した.このノックアウトマウスは若齢においては正常な耐糖能を示したが,加齢にともない24週齢では耐糖能の障害,インスリンの分泌の低下,膵β細胞の量の減少を示し,有糸分裂している膵β細胞および増殖している膵β細胞の有意な低下が認められた.また,妊娠15.5日目においても,野生型のマウスと比較して膵β細胞の量,膵β細胞の有糸分裂および増殖が有意に低下した.
膵β細胞に特異的なCENP-Aノックアウトマウスは雄および雌とも16週間の高脂肪食の負荷により,血糖値の上昇,耐糖能の障害,インスリンの分泌の低下,膵β細胞の量の減少,膵β細胞の有糸分裂および増殖の低下が認められた.また,膵β細胞に特異的なCENP-Aノックアウトマウスの膵島においてはグルコキナーゼ活性化剤による膵β細胞の増殖が有意に抑制された.インスリン受容体のアンタゴニストの投与による急性インスリン抵抗性モデルにおいても,膵β細胞に特異的なCENP-Aノックアウトマウスは血清中のインスリンの濃度,膵β細胞の量,膵β細胞の有糸分裂および増殖の有意な低下が認められた.また,野生型のマウスにインスリン受容体のアンタゴニストを投与したところ,膵島において特異的にCENP-AおよびPLK1の発現が上昇したが,肝臓や脂肪組織において変化はなかった.
野生型の膵β細胞,IRS1を欠損した膵β細胞,IRS2を欠損した膵β細胞においてCENP-Aをノックダウンしたところ細胞の生存および増殖能は低下したが,インスリン受容体を欠損した膵β細胞においてはCENP-Aのノックダウンによるそれらの低下は有意ではなかった.CENP-Aをノックダウンした膵β細胞においては,G2期/M期の細胞が著明に増加し,また,G2期/M期の制御にかかわるサイクリンB1の発現も有意に低下しG2期/M期における細胞周期の停止が示唆された.膵β細胞の増殖の亢進を呈するob/obマウスおよび肝臓に特異的なインスリン受容体ノックアウトマウス8) の膵島においては,細胞の増殖のマーカーと同様にCENP-Aの発現が有意に上昇した.PLK1をノックダウンした膵β細胞およびPLK1に特異的な阻害剤を添加した野生型の膵β細胞は,CENP-Aをノックダウンした膵β細胞と同様に,細胞の増殖の低下およびG2期/M期の細胞の増加を示した.さらに,CENP-Aをノックダウンした膵β細胞,PLK1をノックダウンした膵β細胞,膵β細胞に特異的なCENP-Aノックアウトマウスの膵島において,膵β細胞のアポトーシスは有意に増加した.
FoxM1の阻害剤を添加した野生型の膵β細胞,インスリン受容体を欠損した膵β細胞,PLK1をノックダウンした膵β細胞においては,野生型の膵β細胞と比較して,CENP-Aのセントロメア領域への結合が低下した.ヒトの膵島においても,PI3キナーゼの阻害剤あるいはPLK1の阻害剤の添加によりCENP-Aのセントロメア領域への結合が阻害された.以上より,膵β細胞においてPLK1はCENP-Aのセントロメア領域への結合に重要な役割をはたすことが示唆された.
この研究においては,膵β細胞の量を増殖の促進により増加させ糖尿病の治療に応用するためには,これまでの多くの研究において注目されてきた細胞周期のG0期からG1期への移行やG1期/S期チェックポイントの制御だけでなく,M期の制御が重要であることが示された.膵β細胞において,インスリンによる刺激によりERKがFoxM1の核への移行を促進し,一方で,PI3キナーゼを介したインスリンシグナルがCdk1およびCdk2を介してFoxM1のCENP-A遺伝子領域およびPLK1遺伝子領域への結合を促進することによりCENP-AおよびPLK1の発現を促進し,結果として,PLK1によりCENP-Aのセントロメア領域への結合が増強され有糸分裂が促進される(図2a).インスリンシグナルの低下した膵β細胞では核においてFoxM1が活性化されず,FoxM1,PLK1,CENP-Aを介する経路の低下により代償性の膵β細胞の増殖が障害される(図2b).2型糖尿病における膵β細胞の量の減少にもこれらの機序が関与する可能性が考えられる.
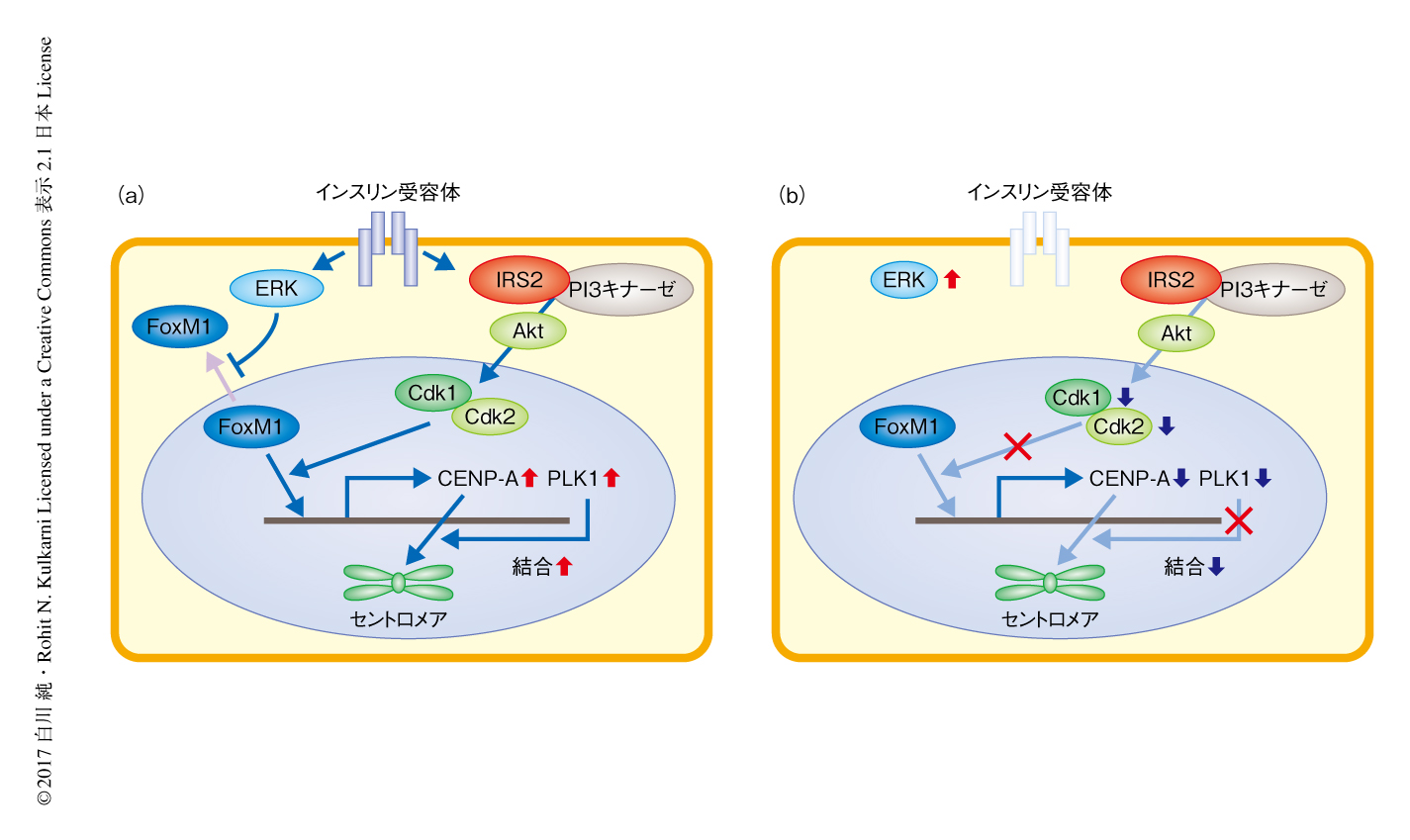
インスリンシグナルを介したCENP-AおよびPLK1の発現の制御は膵β細胞に特異的であり肝臓や脂肪組織においては認められなかったことから,FoxM1,PLK1,CENP-Aを介する経路は高インスリン血症をともなうインスリン抵抗性などにおいて膵β細胞の量のみを特異的に増加させる生体に備わった代償性の機構と想定される.膵β細胞におけるCENP-AおよびPLK1の発現の制御機構として,インスリンにくわえIGF-1やグルコースによる刺激も関与した.また,急性インスリン抵抗性モデルマウスにおいても代償性の膵β細胞の増殖にCENP-Aが必須であった.これらのことにより,代謝障害にともなうインスリン以外の液性因子,脂質,神経性の制御などにより惹起されるFoxM1,PLK1,CENP-Aを介する経路の制御も膵β細胞の代償性の増殖に関与すると考えられる.今後は,このインスリンシグナルに非依存的なCENP-Aを介した膵β細胞の量の制御機構を解明することが課題である.
略歴:2011年 横浜市立大学大学院医学研究科 修了,同年 同 助教,2014年 米国Harvard大学Joslin Diabetes Centerリサーチフェローを経て,2017年より横浜市立大学大学院医学研究科 助教.
研究テーマ:糖尿病および内分泌疾患の基礎研究および臨床研究.
関心事:膵β細胞に着目した糖尿病の治療法.
Rohit N. Kulkarni
米国Harvard大学Joslin Diabetes CenterにてProfessor.
研究室URL:http://www.joslin.org/kulkarni-lab.html
© 2017 白川 純・Rohit N. Kulkarni Licensed under CC 表示 2.1 日本
(米国Harvard大学Joslin Diabetes Center,Section on Islet Cell and Regenerative Biology)
email:白川 純
DOI: 10.7875/first.author.2017.027
Insulin signaling regulates the FoxM1/PLK1/CENP-A pathway to promote adaptive pancreatic β cell proliferation.
Jun Shirakawa, Megan Fernandez, Tomozumi Takatani, Abdelfattah El Ouaamari, Prapaporn Jungtrakoon, Erin R. Okawa, Wei Zhang, Peng Yi, Alessandro Doria, Rohit N. Kulkarni
Cell Metabolism, 25, 868-882.e5 (2017)
要 約
これまで,膵β細胞の増殖の制御についてはおもにG0期からG1期への移行が注目されていたが,この研究においては,M期における染色体の分離に必須のタンパク質であるCENP-Aに着目した.インスリンシグナルは膵β細胞においてCdk1およびCdk2を介して核にて転写因子であるFoxM1を活性化し,CENP-AおよびPLK1の発現を促進した.セントロメア領域へのCENP-Aの結合にはキナーゼであるPLK1が必須であり,CENP-Aをノックダウンした膵β細胞は増殖の低下およびアポトーシスの亢進を示した.膵β細胞に特異的なCENP-Aノックアウトマウスは加齢,妊娠,高脂肪食の負荷,急性インスリン抵抗性モデルにおいて代償性の膵β細胞の増殖が障害された.2型糖尿病の患者に由来する膵島でおいては健常人に由来する膵島と比較して,インスリンシグナルの活性化によるCENP-AおよびPLK1の発現の誘導が低下した.これより,FoxM1,PLK1,CENP-Aを介する経路は代償性の膵β細胞の増殖において必須であり,糖尿病の進行を抑制する治療の標的になりうると考えられた.
はじめに
2型糖尿病においては膵β細胞の量の減少が発症および進展に関与しており,膵β細胞の増殖に着目した治療法が注目されている1).これまで,動物モデルにおいて膵β細胞の増殖あるいは生存にインスリンシグナルが重要であることが報告されている.また,2型糖尿病の患者に由来する膵島においては健常人に由来する膵島と比較してインスリンシグナルに関連するタンパク質の発現が低下することが報告されている2,3).
膵β細胞はもっとも高度に分化した細胞のひとつであり,成体においてほとんどの膵β細胞は静止期すなわちG0期にある.インスリンなどの増殖因子はG0期からG1期への移行の促進やG1期/S期チェックポイントの制御に関与することが報告されているが,それ以降のG2期/M期チェックポイントに関してその制御機構は不明であった(図1).
膵β細胞に特異的なインスリン受容体ノックアウトマウスは,膵β細胞におけるインスリンシグナルの低下により高脂肪食の負荷や肝臓に特異的なインスリン抵抗性モデルにおいて代償性の膵β細胞の増殖が障害される2型糖尿病モデルマウスである4).この研究においては,このノックアウトマウスの膵β細胞において発現の低下したCENP-AおよびPLK1に着目した.
1.インスリンシグナルは膵β細胞においてCENP-AおよびPLK1の発現を制御する
インスリン受容体を欠損した膵β細胞において細胞周期を解析したところ,G0期の細胞およびG1期の細胞が増加しておりG0期における細胞周期の停止が示唆された.G2期の細胞とM期の細胞とを分離したところ,インスリン受容体を欠損した膵β細胞においてM期の細胞が有意に増加しておりM期の進行の低下が示唆された.そこで,膵β細胞に特異的なインスリン受容体ノックアウトマウスの膵β細胞においてマイクロアレイ法を用いて遺伝子発現を網羅的に解析したところ,M期における染色体の分離にかかわるCENP-AおよびM期キナーゼのひとつPLK1の発現が野生型の膵β細胞と比べ有意に低下した.
インスリン受容体を欠損した膵β細胞にインスリンとの結合能の低下した変異型のインスリン受容体を発現させたところ,野生型のインスリン受容体を発現させたときと比べ,CENP-AおよびPLK1の発現は低下した.以上より,膵β細胞においてインスリン受容体へのインスリンの結合がCENP-AおよびPLK1の発現を制御することが示された.転写因子であるFoxM1はCENP-AおよびPLK1の発現を制御することが報告されていたが5,6),インスリン受容体を欠損した膵β細胞およびインスリン受容体を欠損した膵島におけるFoxM1の発現は野生型の膵β細胞と同等であった.膵β細胞においてインスリンのほかにもIGF-1あるいはグルコースによる刺激でもCENP-AおよびPLK1の発現は誘導されたが,グルコースによる刺激はインスリン受容体に依存性であった.
2.インスリンシグナルはCENP-A遺伝子領域およびPLK1遺伝子領域へのFoxM1の結合を促進する
FoxM1の細胞における局在およびDNAへの結合能について解析した.マウスにおいてFoxM1は野生型の膵β細胞およびインスリン受容体を欠損した膵β細胞において,インスリンによる刺激の前後とも核に局在しており,MEK1およびMEK2の阻害剤の添加により核の外へと移行した.ヒトの膵β細胞においてFoxM1は刺激をあたえていない状態ではおもに細胞質に局在してしたが,インスリンによる刺激により核への移行が促進され,MEK1およびMEK2の阻害剤の添加により核への移行が阻害された.インスリン受容体を欠損した膵β細胞においてFoxM1の転写活性への関与が報告されていたCdk1およびCdk2の発現は野生型の膵β細胞と比較して有意に低下した.インスリン受容体およびIGF1受容体の阻害剤,PI3キナーゼの阻害剤,Akt1およびAkt2の阻害剤の存在のもとでは,インスリンの刺激によるCdk1およびCdk2,CENP-A,PLK1の発現の誘導は阻害された.
クロマチン免疫沈降法により,FoxM1はCENP-A遺伝子領域およびPLK1遺伝子領域への結合が示されたが,インスリン受容体を欠損した膵β細胞,また,FoxM1の阻害剤あるいはPI3キナーゼの阻害剤の添加によりその結合能は低下した.これより,膵β細胞においてインスリンシグナルは,FoxM1の発現よりもFoxM1の活性を制御することが示唆された.これらインスリンによるCENP-AおよびPLK1の発現の制御は膵β細胞に特異的であり,肝臓あるいは脂肪組織においてはインスリンによる刺激に非依存性であった.
3.インスリンシグナルによるCENP-Aの発現の誘導は2型糖尿病の患者に由来する膵島において低下する
2型糖尿病の患者に由来する膵島においては,健常人に由来する膵島と比較してmRNAレベルでのCENP-Aの発現は有意に低下した.また,免疫染色によりCENP-A陽性の膵β細胞の割合は2型糖尿病の患者に由来する膵島において有意に低下した.健常人に由来する膵島をインスリンにより刺激したところCENP-AおよびPLK1の発現は有意に上昇したが,2型糖尿病の患者に由来する膵島を刺激しても発現は上昇しなかった.
また,家族性の糖尿病を示す52家系に対する全エクソーム解析により,PLK1遺伝子におけるミスセンス変異が原因遺伝子である可能性が示唆された.
4.膵β細胞に特異的なCENP-Aノックアウトマウスは代償性の膵β細胞の増殖が障害される
MIP-Cre/ERTマウス7) から膵β細胞に特異的なCENP-Aノックアウトマウスを作製した.このノックアウトマウスは若齢においては正常な耐糖能を示したが,加齢にともない24週齢では耐糖能の障害,インスリンの分泌の低下,膵β細胞の量の減少を示し,有糸分裂している膵β細胞および増殖している膵β細胞の有意な低下が認められた.また,妊娠15.5日目においても,野生型のマウスと比較して膵β細胞の量,膵β細胞の有糸分裂および増殖が有意に低下した.
膵β細胞に特異的なCENP-Aノックアウトマウスは雄および雌とも16週間の高脂肪食の負荷により,血糖値の上昇,耐糖能の障害,インスリンの分泌の低下,膵β細胞の量の減少,膵β細胞の有糸分裂および増殖の低下が認められた.また,膵β細胞に特異的なCENP-Aノックアウトマウスの膵島においてはグルコキナーゼ活性化剤による膵β細胞の増殖が有意に抑制された.インスリン受容体のアンタゴニストの投与による急性インスリン抵抗性モデルにおいても,膵β細胞に特異的なCENP-Aノックアウトマウスは血清中のインスリンの濃度,膵β細胞の量,膵β細胞の有糸分裂および増殖の有意な低下が認められた.また,野生型のマウスにインスリン受容体のアンタゴニストを投与したところ,膵島において特異的にCENP-AおよびPLK1の発現が上昇したが,肝臓や脂肪組織において変化はなかった.
5.CENP-AおよびPLK1の膵β細胞の増殖および生存における役割
野生型の膵β細胞,IRS1を欠損した膵β細胞,IRS2を欠損した膵β細胞においてCENP-Aをノックダウンしたところ細胞の生存および増殖能は低下したが,インスリン受容体を欠損した膵β細胞においてはCENP-Aのノックダウンによるそれらの低下は有意ではなかった.CENP-Aをノックダウンした膵β細胞においては,G2期/M期の細胞が著明に増加し,また,G2期/M期の制御にかかわるサイクリンB1の発現も有意に低下しG2期/M期における細胞周期の停止が示唆された.膵β細胞の増殖の亢進を呈するob/obマウスおよび肝臓に特異的なインスリン受容体ノックアウトマウス8) の膵島においては,細胞の増殖のマーカーと同様にCENP-Aの発現が有意に上昇した.PLK1をノックダウンした膵β細胞およびPLK1に特異的な阻害剤を添加した野生型の膵β細胞は,CENP-Aをノックダウンした膵β細胞と同様に,細胞の増殖の低下およびG2期/M期の細胞の増加を示した.さらに,CENP-Aをノックダウンした膵β細胞,PLK1をノックダウンした膵β細胞,膵β細胞に特異的なCENP-Aノックアウトマウスの膵島において,膵β細胞のアポトーシスは有意に増加した.
6.膵β細胞におけるPLK1に依存的なCENP-Aのセントロメア領域への結合
FoxM1の阻害剤を添加した野生型の膵β細胞,インスリン受容体を欠損した膵β細胞,PLK1をノックダウンした膵β細胞においては,野生型の膵β細胞と比較して,CENP-Aのセントロメア領域への結合が低下した.ヒトの膵島においても,PI3キナーゼの阻害剤あるいはPLK1の阻害剤の添加によりCENP-Aのセントロメア領域への結合が阻害された.以上より,膵β細胞においてPLK1はCENP-Aのセントロメア領域への結合に重要な役割をはたすことが示唆された.
おわりに
この研究においては,膵β細胞の量を増殖の促進により増加させ糖尿病の治療に応用するためには,これまでの多くの研究において注目されてきた細胞周期のG0期からG1期への移行やG1期/S期チェックポイントの制御だけでなく,M期の制御が重要であることが示された.膵β細胞において,インスリンによる刺激によりERKがFoxM1の核への移行を促進し,一方で,PI3キナーゼを介したインスリンシグナルがCdk1およびCdk2を介してFoxM1のCENP-A遺伝子領域およびPLK1遺伝子領域への結合を促進することによりCENP-AおよびPLK1の発現を促進し,結果として,PLK1によりCENP-Aのセントロメア領域への結合が増強され有糸分裂が促進される(図2a).インスリンシグナルの低下した膵β細胞では核においてFoxM1が活性化されず,FoxM1,PLK1,CENP-Aを介する経路の低下により代償性の膵β細胞の増殖が障害される(図2b).2型糖尿病における膵β細胞の量の減少にもこれらの機序が関与する可能性が考えられる.
インスリンシグナルを介したCENP-AおよびPLK1の発現の制御は膵β細胞に特異的であり肝臓や脂肪組織においては認められなかったことから,FoxM1,PLK1,CENP-Aを介する経路は高インスリン血症をともなうインスリン抵抗性などにおいて膵β細胞の量のみを特異的に増加させる生体に備わった代償性の機構と想定される.膵β細胞におけるCENP-AおよびPLK1の発現の制御機構として,インスリンにくわえIGF-1やグルコースによる刺激も関与した.また,急性インスリン抵抗性モデルマウスにおいても代償性の膵β細胞の増殖にCENP-Aが必須であった.これらのことにより,代謝障害にともなうインスリン以外の液性因子,脂質,神経性の制御などにより惹起されるFoxM1,PLK1,CENP-Aを介する経路の制御も膵β細胞の代償性の増殖に関与すると考えられる.今後は,このインスリンシグナルに非依存的なCENP-Aを介した膵β細胞の量の制御機構を解明することが課題である.
文 献
- Shirakawa, J. & Kulkarni, R. N.: Novel factors modulating human β-cell proliferation. Diabetes Obes. Metab., 18 (Suppl. 1), 71-77 (2016)[PubMed]
- Folli, F., Okada, T., Perego, C. et al.: Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS One, 6, e28050 (2011)[PubMed]
- Gunton, J. E., Kulkarni, R. N., Yim, S. et al.: Loss of ARNT/HIF1β mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell, 122, 337-349 (2005)[PubMed]
- Kulkarni, R. N., Bruning, J. C., Winnay, J. N. et al.: Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell, 96, 329-339 (1999)[PubMed]
- Golson, M. L., Dunn, J. C., Maulis, M. F. et al.: Activation of FoxM1 revitalizes the replicative potential of aged β-cells in male mice and enhances insulin secretion. Diabetes, 64, 3829-3838 (2015)[PubMed]
- Wang, I. C., Chen, Y. J., Hughes, D. et al.: Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell. Biol., 25, 10875-10894 (2005)[PubMed]
- Wicksteed, B., Brissova, M., Yan, W. et al.: Conditional gene targeting in mouse pancreatic β-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes, 59, 3090-3098 (2010)[PubMed]
- Michael, M. D., Kulkarni, R. N., Postic, C. et al.: Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell, 6, 87-97 (2000)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2011年 横浜市立大学大学院医学研究科 修了,同年 同 助教,2014年 米国Harvard大学Joslin Diabetes Centerリサーチフェローを経て,2017年より横浜市立大学大学院医学研究科 助教.
研究テーマ:糖尿病および内分泌疾患の基礎研究および臨床研究.
関心事:膵β細胞に着目した糖尿病の治療法.
Rohit N. Kulkarni
米国Harvard大学Joslin Diabetes CenterにてProfessor.
研究室URL:http://www.joslin.org/kulkarni-lab.html
© 2017 白川 純・Rohit N. Kulkarni Licensed under CC 表示 2.1 日本
