異質4倍体であるアフリカツメガエルにおけるゲノムの進化
宇野好宣1・平良眞規2
(1名古屋大学大学院生命農学研究科 応用分子生命科学専攻動物遺伝制御学研究室,2東京大学大学院理学系研究科 生物科学専攻生物学講座分子生物学研究室)
email:宇野好宣,平良眞規
DOI: 10.7875/first.author.2016.125
Genome evolution in the allotetraploid frog Xenopus laevis.
Adam M. Session, Yoshinobu Uno, Taejoon Kwon, Jarrod A. Chapman, Atsushi Toyoda, Shuji Takahashi, Akimasa Fukui, Akira Hikosaka, Atsushi Suzuki, Mariko Kondo, Simon J. van Heeringen, Ian Quigley, Sven Heinz, Hajime Ogino, Haruki Ochi, Uffe Hellsten, Jessica B. Lyons, Oleg Simakov, Nicholas Putnam, Jonathan Stites, Yoko Kuroki, Toshiaki Tanaka, Tatsuo Michiue, Minoru Watanabe, Ozren Bogdanovic, Ryan Lister, Georgios Georgiou, Sarita S. Paranjpe, Ila van Kruijsbergen, Shengquiang Shu, Joseph Carlson, Tsutomu Kinoshita, Yuko Ohta, Shuuji Mawaribuchi, Jerry Jenkins, Jane Grimwood, Jeremy Schmutz, Therese Mitros, Sahar V. Mozaffari, Yutaka Suzuki, Yoshikazu Haramoto, Takamasa S. Yamamoto, Chiyo Takagi, Rebecca Heald, Kelly Miller, Christian Haudenschild, Jacob Kitzman, Takuya Nakayama, Yumi Izutsu, Jacques Robert, Joshua Fortriede, Kevin Burns, Vaneet Lotay, Kamran Karimi, Yuuri Yasuoka, Darwin S. Dichmann, Martin F. Flajnik, Douglas W. Houston, Jay Shendure, Louis DuPasquier, Peter D. Vize, Aaron M. Zorn, Michihiko Ito, Edward M. Marcotte, John B. Wallingford, Yuzuru Ito, Makoto Asashima, Naoto Ueno, Yoichi Matsuda, Gert Jan C. Veenstra, Asao Fujiyama, Richard M. Harland, Masanori Taira, Daniel S. Rokhsar
Nature, 538, 336-343 (2016)
非常に有用なモデル生物として古くから用いられているアフリカツメガエルは,異種交配および全ゲノム重複により,ひとつの生物のなかに異なる2種類のゲノムをもつ異質4倍体と考えられていた.そのため,全ゲノム配列の解読は非常に困難とあきらめられており,主要なモデル生物のなかで唯一,全ゲノム配列が明らかにされていなかった.筆者らは,アフリカツメガエルの全ゲノム配列の解読に挑み,6年をかけてついにその構造を明らかにした.その結果,アフリカツメガエルは約1800万年前に2つの祖先種の異種交配により誕生した異質4倍体種であり,祖先種からうけついだ2つのサブゲノムは,それらのあいだで相互転座など構造異常の起こることなく,それぞれ9本の染色体のセットに分かれて保持されていることが明らかにされた.さらに,一方のサブゲノムにおいては多くの重複遺伝子の欠失,発現量の低下,高頻度な染色体の再配列がみられたことから,サブゲノムごとに独自に進化したことが明らかにされた.
今日,多くの生物種において全ゲノム配列が明らかにされており,得られたゲノム情報を用いた比較ゲノム解析や遺伝子の網羅的な機能解析などにより多くの知見がもたらされている.これまで,脊椎動物の全ゲノム配列は,最初にヒトにおいて明らかにされたのち,マウス,ニワトリ,ゼブラフィッシュ,メダカなど世界的に多くの研究者に用いられている主要なモデル生物を中心として明らかにされてきた.アフリカツメガエル(Xenopus laevis)は,1950年代から現在にいたるまで,発生学,細胞生物学,生化学などにおいて有用なモデル生物として多くの研究者に利用されてきた(図1).しかしながら,研究の歴史が古い主要なモデル生物としては唯一,全ゲノム配列が明らかにされていなかった.

多くの生物種は父方および母方からそれぞれうけついだ2つの同一のゲノムをもつ2倍体であるが,アフリカツメガエルは2つの2倍体の祖先種の異種交配および全ゲノム重複により生じた,異なる2つのサブゲノムをもつ異質4倍体種と考えられていた1).この異種交配を起こした2倍体の祖先種はすでに絶滅しているので,アフリカツメガエルのゲノムの比較の対象としては,約4800万年前に分岐した近縁種である2倍体のネッタイツメガエルがある(図1).2010年,両生類としてはじめてネッタイツメガエルの全ゲノム配列が明らかにされたが2),異質4倍体のアフリカツメガエルは相同性の高い2つの祖先種に由来するサブゲノムをもつことから,全ゲノム配列の解読は困難と考えられていた.しかし,この研究において,日本および米国を中心とするアフリカツメガエル全ゲノム解読プロジェクトの国際コンソーシアムは,異なる2つの祖先種に由来する同祖染色体および2つの祖先種のオーソログに由来する同祖遺伝子を識別して全ゲノム配列を解読することにより,複雑な異質4倍数体のゲノムの構造を明らかにした.さらに,祖先種に由来する2つのサブゲノムの同定により,倍数体の動物種においてはじめてサブゲノムごとの進化の過程の一端が解明された.
アフリカツメガエル全ゲノム解読プロジェクトは,2009年に日本および米国において独立にたちあがったが,2012年,国際コンソーシアムとして共同することで合意した.それを可能にしたのは,日本が独自に作出したアフリカツメガエル唯一の近交系であるJ系統を両方のチームで用いていたことである.J系統はゲノム配列に個体差がないため,2つの祖先種に由来するサブゲノムの配列の違いをうかびあがらせることが可能である.米国チームは,J系統の個体のゲノムDNAを用いてショットガンシークエンスを行い,その配列をつなぎあわせてアセンブル配列を得た.ついで,メイトペアシークエンスを行い,その配列をもとにアセンブル配列をつなぎスカフォールド配列を構築した.しかし,これだけではよく似た2つのサブゲノムに由来するアセンブル配列やスカフォールド配列が正しく結合しているかどうかは不確実である.日本チームは,BAC(bacterial artificial chromosome,細菌人工染色体)クローンライブラリーを作製し,その末端配列を決定してスカフォールド配列にマッピングした.ついで,マッピングされたBACクローンをプローブとして蛍光in situハイブリダイゼーション法を用いて染色体にマッピングし,アフリカツメガエルの18対すべての染色体を識別しながら,スカフォールド配列にマッピングされたBACクローンをそれぞれの染色体のアンカーマーカーとして設定した.最終的には,798個のBACクローンからなる染色体地図が作成された.一方,米国チームも,Hi-C法3) やChicago法4) により染色体のレベルでアセンブリーを進め,BACクローンの蛍光in situハイブリダイゼーション法の結果と照合しながら,染色体にアンカーされていないスカフォールド配列も含め,ほとんどの配列を染色体ごとのひとつづきの配列としてつないだ.
しかし,配列の自動アセンブリーには限界があり,たとえば,遺伝子が直列に重複したところなどはスカフォールド配列がとぎれたり1個に縮重していたりした.そのような箇所では,それをまたぐようなBACクローンを選別しその全塩基配列を決定した.その結果,Hox遺伝子クラスター,嗅覚受容体遺伝子クラスター,nodal5遺伝子クラスター,nodal3遺伝子クラスター,vg1遺伝子クラスターなどの配列が決定された.とくに,vg1遺伝子はクラスターを形成することがはじめて示され,その結果,これまでvg1遺伝子のバリアントと考えられていたvg1(S20) 遺伝子とvg1(P20) 遺伝子はひとつのクラスターのなかの別々の遺伝子であることが判明した.これらの配列も含めて,18本の染色体ごとの配列はver. 9.1として公開された(Xenbase,および,
http://xenopus.lab.nig.ac.jp/cgi-bin/gb2/gbrowse/xl_v9_1m_p/).
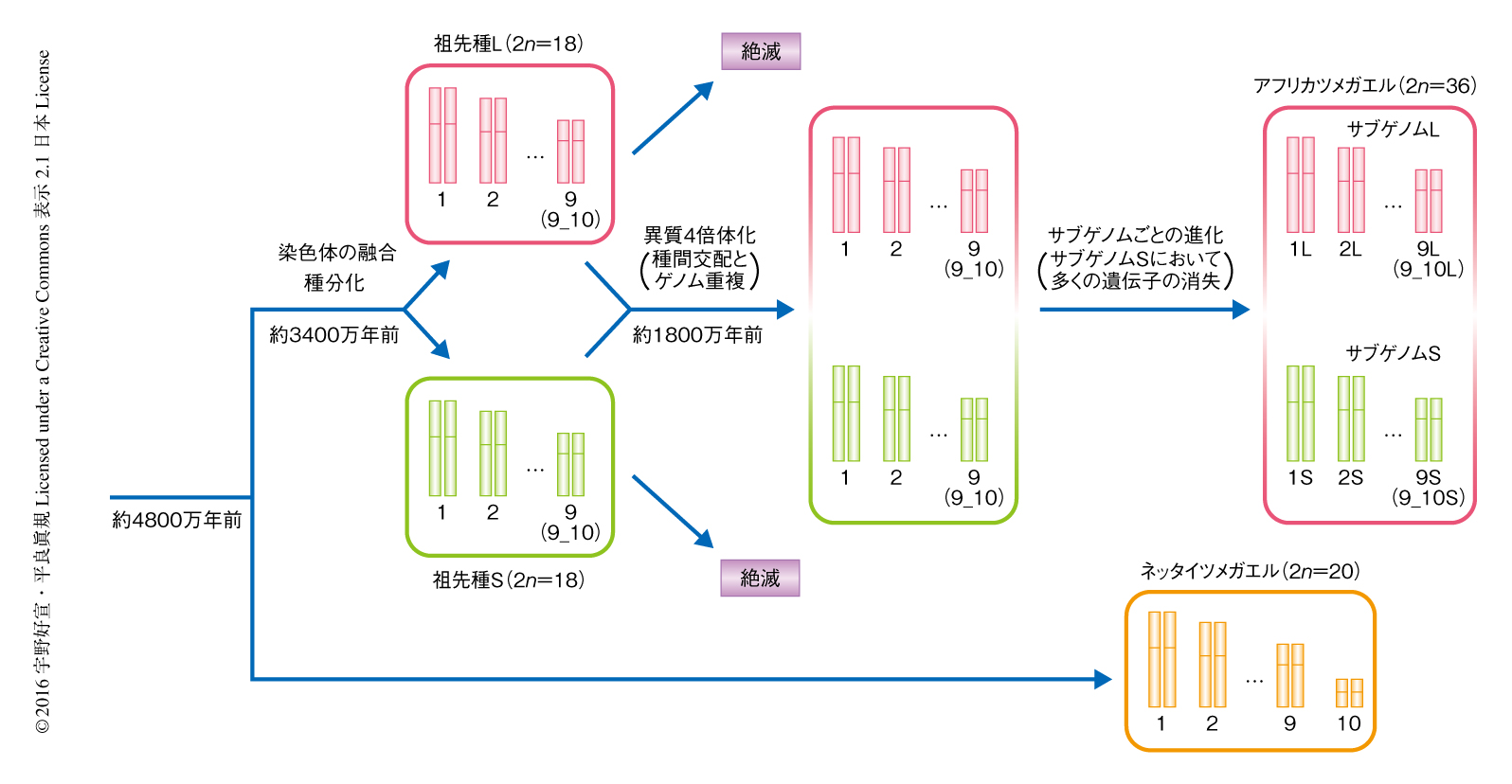
今回の解析をもとに,これまで不統一であったアフリカツメガエルの染色体の番号が新たに設定しなおされた5).つまり,ネッタイツメガエルの染色体(XTRと表記)のXTR1からXTR10と,アフリカツメガエルの9対の同祖染色体における遺伝子連鎖群の保存性6) をもとに,アフリカツメガエルの染色体(XLAと表記)の番号をふりなおし,かつ,同祖染色体のあいだの長さの違いをもとに長い方をL,短い方をSと表記して,結果として,XLA1L,XLA1S,XLA2L,XLA2S,… と命名した.ただし,ネッタイツメガエルのXTR9およびXTR10と相同な染色体はアフリカツメガエルにおいて融合していたことから,XLA9_10LおよびXLA9_10S(あるいは,XLA9LおよびXLA9S)とした.XLA9_10の融合領域は,ネッタイツメガエルのXTR9およびXTR10の末端の領域の配列との詳細な比較により,XLA9_10LおよびXLA9_10Sに存在する4つの遺伝子を含む領域にまでせばめられた.これらの遺伝子はネッタイツメガエルのXTR9あるいはXTR10には含まれていなかったため,蛍光in situハイブリダイゼーション法により,実際にXTR9およびXTR10の末端の領域に存在することを示し融合領域を同定した.XLA9_10LとXLA9_10Sの融合領域は同一であったことから,融合が起こったのはネッタイツメガエルから分岐したあとで,かつ,アフリカツメガエルの2つの2倍体の祖先種が種分化するまえであると考えられた(図2).
ゲノムの配列をもとにしたタンパク質をコードする遺伝子モデルの作成および遺伝子のアノテーションは,米国チームにより自動化ツールを用いて行われた.しかし,自動化ツールにより作成された遺伝子モデルにおいては,しばしば,エクソン-イントロン構造にまちがいが含まれたり,遺伝子名の付与が適切でなかったりする場合がある.そこで,日本チームにより,シグナル伝達,転写制御,細胞周期,主要組織適合性複合体などに関連する遺伝子を中心に検定を行った.エクソン-イントロンが本来の位置に設定されていなかったものをリストアップし,それが修正されるよう自動化ツールの条件を設定しなおし,かつ,この作業を何度もくり返すことにより正確さを向上させた.それと並行して,遺伝子のアノテーションの検定も行い,987個の遺伝子について検定し修正すべきものは修正した.これらの結果を反映させて,遺伝子アノテーションver. 1.8として,ゲノム配列とともに公開した.
アフリカツメガエルのもつ18対の染色体がLとSの9対の同祖染色体となることはすでに述べた5).しかし,祖先種に由来する2つのサブゲノムが同祖染色体のあいだでどのように分布しているのか,また,同祖染色体のあいだで組換えが起こっているのかどうかは不明であった.そこで,注目したのがトランスポゾンである.もし,2種の祖先種がそれぞれの種に特異的なトランスポゾンをもつのであれば,異質4倍体化したアフリカツメガエルにおいてそのトランスポゾンの配列がそれぞれのサブゲノムの全域に保持されているはずである.そこで,すでに不活性化して“化石化”しているトランスポゾンに注目し,同祖染色体のあいだに不均等に分布しているものはないか探索した.その結果,3種類の化石化DNAトランスポゾンとしてXl-TpL_harb,Xl-TpS_harb,Xl-TpS_Marが見い出された.そのうちの1つXl-TpL_harbは染色体Lのセットに特異的に分布していた一方,ほかの2つXl-TpS_harbおよびXl-TpS_Marは染色体Sのセットに特異的に分布していた.つまり,アフリカツメガエルの染色体Lと染色体Sのセットはサブゲノムに対応していることが明らかにされた.そこで,染色体Lと染色体Sに対応させてサブゲノムLとサブゲノムSと命名し,それらの起源となる絶滅した祖先種もLとSとした.また,それらのサブゲノムに属する同祖遺伝子は遺伝子名のあとに“.L”あるいは“.S”を付加して表記することにした.倍数体の動物種におけるサブゲノムの同定ははじめてのことであった.これにより,染色体Lと染色体Sに存在する遺伝子を調べることにより,異質倍数体化したのちのサブゲノムLとサブゲノムSの進化を解析することが可能になった.
アフリカツメガエルにおいてサブゲノムのあいだの比較が可能になったことから,近縁種のネッタイツメガエルとアフリカツメガエルの2つのサブゲノムにおいて詳細な比較解析を行った.その結果,アフリカツメガエルの染色体のセットLはネッタイツメガエルの染色体とシンテニーや染色体の形態が非常に類似していたが,染色体のセットSは染色体のセットLと比べ染色体の長さが短く,染色体のレベルでの構造変化が多く生じていた.2倍体のネッタイツメガエルの遺伝子数は約21,000個であるが,アフリカツメガエルの遺伝子数はその約2倍の45,099個であった.これらのうち,嗅覚受容体遺伝子ファミリーのように多コピーで存在している遺伝子などを除いた24,419個の遺伝子のうち,異質倍数化により重複した2コピーが保存されている遺伝子は全体の56%である8806ペア(17,612遺伝子)であり,1コピーが欠失した遺伝子は6807遺伝子であった.そこで,染色体のセットごとの遺伝子の保存性について調べた結果,異質倍数化したのちサブゲノムLでは8.3%の遺伝子が消失していたのに対し,サブゲノムSでは31.5%の遺伝子が消失していた.
さらに,遺伝子の発現パターンや発現量についても比較した.14の異なる段階の卵母細胞あるいは発生段階の胚および14の異なる成体の組織を用いたRNA-Seq法により,大量のcDNAの配列情報を得た.これらをゲノム配列にマップして遺伝子の発現情報を得た.その結果,卵母細胞から母性-胚性転移期の胚にかけてはサブゲノムLの遺伝子の発現量がサブゲノムSの遺伝子より平均で12%高かったのに対し,母性-胚性転移期よりのちの胚および成体組織においてはサブゲノムLの遺伝子の発現量はサブゲノムSの遺伝子より25%高いことが明らかにされた.これらの結果から,アフリカツメガエルにおいては,1800万年前に祖先種Lと祖先種Sの種間交配およびゲノム重複による異質倍数化が起こり,そののち,サブゲノムLでは祖先種Lのゲノムをほぼそのまま残してきたのに対し,サブゲノムSでは多くの遺伝子の消失,偽遺伝子化,高頻度な染色体の再配列,そして,多くの遺伝子の発現量の低下が発生過程の胚および成体において起こったことが明らかにされた(図2).トウモロコシや出芽酵母の異質倍数体種においては,一方のサブゲノムの染色体領域の一部において多くの重複遺伝子の消失が起こっていると報告されている7,8).しかしながら,アフリカツメガエルはこれらとは異なり,サブゲノムSの全域において遺伝子の消失や染色体の再配列が生じていた.
全ゲノム重複は生物の進化の過程においてしばしば起こる現象と考えられており,そのひとつの例が,約5億年前の古生代カンブリア紀に脊椎動物が出現する過程で起こったとされる2回の全ゲノム重複である9).全ゲノム重複により遺伝子の数を格段に増加させ,一部の重複遺伝子の機能の分担や新たな機能の獲得が脊椎動物の誕生とそののちの多様化および繁栄をもたらした要因であったと考えられている10).また,真骨魚類の祖先種において約3億年前に独自に全ゲノム重複が起こり,さらに,最近,全ゲノム配列が明らかにされたニジマスにおいては約1億年前にもう1回の全ゲノム重複が起こっている11).しかしながら,これらの全ゲノム重複からはいずれも1億年以上が経過しているため,祖先種に由来するサブゲノムを明らかにすることができず,サブゲノムごとの進化の過程を推測するのは困難であった.この研究においては,約1800万年前という比較的最近に全ゲノム重複の起こったアフリカツメガエルの全ゲノム配列を解読することにより,異質倍数体の動物種においてはじめてサブゲノムを識別することに成功し,それをもとに,全ゲノム重複ののちのサブゲノムの進化がはじめて明らかにされた.さらに,アフリカツメガエルと同じツメガエル属には,さらに1回あるいは2回の全ゲノム重複が想定される種が存在する12).これらの倍数体のツメガエルにおいてゲノムの構造を明らかにしアフリカツメガエルとの比較解析を行うことは,約5億年前に起こったとされる脊椎動物の共通祖先で生じた2回の全ゲノム重複や,そののち,真骨魚類に起こった全ゲノム重複が,のちの進化にどのようなインパクトをあたえたかを読み解く鍵,すなわち,ロゼッタストーンになるものと期待される.
この研究の成果はNature誌に掲載されただけでなく,このプロジェクトの日本チームにより8報のcompanion paperがDevelopmental Biology誌に掲載された13-20).
略歴:2011年 北海道大学大学院生命科学院 修了,同年より名古屋大学大学院生命農学研究科 研究員.
研究テーマ:脊椎動物のゲノムおよび染色体の進化.
抱負:この研究におけるアフリカツメガエルの全ゲノム重複のように,ある生物種のゲノムや染色体を詳細に調べることにより,脊椎動物における進化の謎の一端を解明したい.
平良 眞規(Masanori Taira)
東京大学大学院理学系研究科 准教授.
研究室URL:http://www.biol.s.u-tokyo.ac.jp/users/lmb/lmb-hp.html
© 2016 宇野好宣・平良眞規 Licensed under CC 表示 2.1 日本
(1名古屋大学大学院生命農学研究科 応用分子生命科学専攻動物遺伝制御学研究室,2東京大学大学院理学系研究科 生物科学専攻生物学講座分子生物学研究室)
email:宇野好宣,平良眞規
DOI: 10.7875/first.author.2016.125
Genome evolution in the allotetraploid frog Xenopus laevis.
Adam M. Session, Yoshinobu Uno, Taejoon Kwon, Jarrod A. Chapman, Atsushi Toyoda, Shuji Takahashi, Akimasa Fukui, Akira Hikosaka, Atsushi Suzuki, Mariko Kondo, Simon J. van Heeringen, Ian Quigley, Sven Heinz, Hajime Ogino, Haruki Ochi, Uffe Hellsten, Jessica B. Lyons, Oleg Simakov, Nicholas Putnam, Jonathan Stites, Yoko Kuroki, Toshiaki Tanaka, Tatsuo Michiue, Minoru Watanabe, Ozren Bogdanovic, Ryan Lister, Georgios Georgiou, Sarita S. Paranjpe, Ila van Kruijsbergen, Shengquiang Shu, Joseph Carlson, Tsutomu Kinoshita, Yuko Ohta, Shuuji Mawaribuchi, Jerry Jenkins, Jane Grimwood, Jeremy Schmutz, Therese Mitros, Sahar V. Mozaffari, Yutaka Suzuki, Yoshikazu Haramoto, Takamasa S. Yamamoto, Chiyo Takagi, Rebecca Heald, Kelly Miller, Christian Haudenschild, Jacob Kitzman, Takuya Nakayama, Yumi Izutsu, Jacques Robert, Joshua Fortriede, Kevin Burns, Vaneet Lotay, Kamran Karimi, Yuuri Yasuoka, Darwin S. Dichmann, Martin F. Flajnik, Douglas W. Houston, Jay Shendure, Louis DuPasquier, Peter D. Vize, Aaron M. Zorn, Michihiko Ito, Edward M. Marcotte, John B. Wallingford, Yuzuru Ito, Makoto Asashima, Naoto Ueno, Yoichi Matsuda, Gert Jan C. Veenstra, Asao Fujiyama, Richard M. Harland, Masanori Taira, Daniel S. Rokhsar
Nature, 538, 336-343 (2016)
この論文に出現する遺伝子・タンパク質のUniprot ID
Hox, 嗅覚受容体, nodal5, nodal3, vg1, S20, P20
要 約
非常に有用なモデル生物として古くから用いられているアフリカツメガエルは,異種交配および全ゲノム重複により,ひとつの生物のなかに異なる2種類のゲノムをもつ異質4倍体と考えられていた.そのため,全ゲノム配列の解読は非常に困難とあきらめられており,主要なモデル生物のなかで唯一,全ゲノム配列が明らかにされていなかった.筆者らは,アフリカツメガエルの全ゲノム配列の解読に挑み,6年をかけてついにその構造を明らかにした.その結果,アフリカツメガエルは約1800万年前に2つの祖先種の異種交配により誕生した異質4倍体種であり,祖先種からうけついだ2つのサブゲノムは,それらのあいだで相互転座など構造異常の起こることなく,それぞれ9本の染色体のセットに分かれて保持されていることが明らかにされた.さらに,一方のサブゲノムにおいては多くの重複遺伝子の欠失,発現量の低下,高頻度な染色体の再配列がみられたことから,サブゲノムごとに独自に進化したことが明らかにされた.
はじめに
今日,多くの生物種において全ゲノム配列が明らかにされており,得られたゲノム情報を用いた比較ゲノム解析や遺伝子の網羅的な機能解析などにより多くの知見がもたらされている.これまで,脊椎動物の全ゲノム配列は,最初にヒトにおいて明らかにされたのち,マウス,ニワトリ,ゼブラフィッシュ,メダカなど世界的に多くの研究者に用いられている主要なモデル生物を中心として明らかにされてきた.アフリカツメガエル(Xenopus laevis)は,1950年代から現在にいたるまで,発生学,細胞生物学,生化学などにおいて有用なモデル生物として多くの研究者に利用されてきた(図1).しかしながら,研究の歴史が古い主要なモデル生物としては唯一,全ゲノム配列が明らかにされていなかった.
多くの生物種は父方および母方からそれぞれうけついだ2つの同一のゲノムをもつ2倍体であるが,アフリカツメガエルは2つの2倍体の祖先種の異種交配および全ゲノム重複により生じた,異なる2つのサブゲノムをもつ異質4倍体種と考えられていた1).この異種交配を起こした2倍体の祖先種はすでに絶滅しているので,アフリカツメガエルのゲノムの比較の対象としては,約4800万年前に分岐した近縁種である2倍体のネッタイツメガエルがある(図1).2010年,両生類としてはじめてネッタイツメガエルの全ゲノム配列が明らかにされたが2),異質4倍体のアフリカツメガエルは相同性の高い2つの祖先種に由来するサブゲノムをもつことから,全ゲノム配列の解読は困難と考えられていた.しかし,この研究において,日本および米国を中心とするアフリカツメガエル全ゲノム解読プロジェクトの国際コンソーシアムは,異なる2つの祖先種に由来する同祖染色体および2つの祖先種のオーソログに由来する同祖遺伝子を識別して全ゲノム配列を解読することにより,複雑な異質4倍数体のゲノムの構造を明らかにした.さらに,祖先種に由来する2つのサブゲノムの同定により,倍数体の動物種においてはじめてサブゲノムごとの進化の過程の一端が解明された.
1.異質4倍体であるアフリカツメガエルの全ゲノム解読および染色体へのマッピング
アフリカツメガエル全ゲノム解読プロジェクトは,2009年に日本および米国において独立にたちあがったが,2012年,国際コンソーシアムとして共同することで合意した.それを可能にしたのは,日本が独自に作出したアフリカツメガエル唯一の近交系であるJ系統を両方のチームで用いていたことである.J系統はゲノム配列に個体差がないため,2つの祖先種に由来するサブゲノムの配列の違いをうかびあがらせることが可能である.米国チームは,J系統の個体のゲノムDNAを用いてショットガンシークエンスを行い,その配列をつなぎあわせてアセンブル配列を得た.ついで,メイトペアシークエンスを行い,その配列をもとにアセンブル配列をつなぎスカフォールド配列を構築した.しかし,これだけではよく似た2つのサブゲノムに由来するアセンブル配列やスカフォールド配列が正しく結合しているかどうかは不確実である.日本チームは,BAC(bacterial artificial chromosome,細菌人工染色体)クローンライブラリーを作製し,その末端配列を決定してスカフォールド配列にマッピングした.ついで,マッピングされたBACクローンをプローブとして蛍光in situハイブリダイゼーション法を用いて染色体にマッピングし,アフリカツメガエルの18対すべての染色体を識別しながら,スカフォールド配列にマッピングされたBACクローンをそれぞれの染色体のアンカーマーカーとして設定した.最終的には,798個のBACクローンからなる染色体地図が作成された.一方,米国チームも,Hi-C法3) やChicago法4) により染色体のレベルでアセンブリーを進め,BACクローンの蛍光in situハイブリダイゼーション法の結果と照合しながら,染色体にアンカーされていないスカフォールド配列も含め,ほとんどの配列を染色体ごとのひとつづきの配列としてつないだ.
しかし,配列の自動アセンブリーには限界があり,たとえば,遺伝子が直列に重複したところなどはスカフォールド配列がとぎれたり1個に縮重していたりした.そのような箇所では,それをまたぐようなBACクローンを選別しその全塩基配列を決定した.その結果,Hox遺伝子クラスター,嗅覚受容体遺伝子クラスター,nodal5遺伝子クラスター,nodal3遺伝子クラスター,vg1遺伝子クラスターなどの配列が決定された.とくに,vg1遺伝子はクラスターを形成することがはじめて示され,その結果,これまでvg1遺伝子のバリアントと考えられていたvg1(S20) 遺伝子とvg1(P20) 遺伝子はひとつのクラスターのなかの別々の遺伝子であることが判明した.これらの配列も含めて,18本の染色体ごとの配列はver. 9.1として公開された(Xenbase,および,
http://xenopus.lab.nig.ac.jp/cgi-bin/gb2/gbrowse/xl_v9_1m_p/).
今回の解析をもとに,これまで不統一であったアフリカツメガエルの染色体の番号が新たに設定しなおされた5).つまり,ネッタイツメガエルの染色体(XTRと表記)のXTR1からXTR10と,アフリカツメガエルの9対の同祖染色体における遺伝子連鎖群の保存性6) をもとに,アフリカツメガエルの染色体(XLAと表記)の番号をふりなおし,かつ,同祖染色体のあいだの長さの違いをもとに長い方をL,短い方をSと表記して,結果として,XLA1L,XLA1S,XLA2L,XLA2S,… と命名した.ただし,ネッタイツメガエルのXTR9およびXTR10と相同な染色体はアフリカツメガエルにおいて融合していたことから,XLA9_10LおよびXLA9_10S(あるいは,XLA9LおよびXLA9S)とした.XLA9_10の融合領域は,ネッタイツメガエルのXTR9およびXTR10の末端の領域の配列との詳細な比較により,XLA9_10LおよびXLA9_10Sに存在する4つの遺伝子を含む領域にまでせばめられた.これらの遺伝子はネッタイツメガエルのXTR9あるいはXTR10には含まれていなかったため,蛍光in situハイブリダイゼーション法により,実際にXTR9およびXTR10の末端の領域に存在することを示し融合領域を同定した.XLA9_10LとXLA9_10Sの融合領域は同一であったことから,融合が起こったのはネッタイツメガエルから分岐したあとで,かつ,アフリカツメガエルの2つの2倍体の祖先種が種分化するまえであると考えられた(図2).
ゲノムの配列をもとにしたタンパク質をコードする遺伝子モデルの作成および遺伝子のアノテーションは,米国チームにより自動化ツールを用いて行われた.しかし,自動化ツールにより作成された遺伝子モデルにおいては,しばしば,エクソン-イントロン構造にまちがいが含まれたり,遺伝子名の付与が適切でなかったりする場合がある.そこで,日本チームにより,シグナル伝達,転写制御,細胞周期,主要組織適合性複合体などに関連する遺伝子を中心に検定を行った.エクソン-イントロンが本来の位置に設定されていなかったものをリストアップし,それが修正されるよう自動化ツールの条件を設定しなおし,かつ,この作業を何度もくり返すことにより正確さを向上させた.それと並行して,遺伝子のアノテーションの検定も行い,987個の遺伝子について検定し修正すべきものは修正した.これらの結果を反映させて,遺伝子アノテーションver. 1.8として,ゲノム配列とともに公開した.
2.祖先種に由来するサブゲノムの同定
アフリカツメガエルのもつ18対の染色体がLとSの9対の同祖染色体となることはすでに述べた5).しかし,祖先種に由来する2つのサブゲノムが同祖染色体のあいだでどのように分布しているのか,また,同祖染色体のあいだで組換えが起こっているのかどうかは不明であった.そこで,注目したのがトランスポゾンである.もし,2種の祖先種がそれぞれの種に特異的なトランスポゾンをもつのであれば,異質4倍体化したアフリカツメガエルにおいてそのトランスポゾンの配列がそれぞれのサブゲノムの全域に保持されているはずである.そこで,すでに不活性化して“化石化”しているトランスポゾンに注目し,同祖染色体のあいだに不均等に分布しているものはないか探索した.その結果,3種類の化石化DNAトランスポゾンとしてXl-TpL_harb,Xl-TpS_harb,Xl-TpS_Marが見い出された.そのうちの1つXl-TpL_harbは染色体Lのセットに特異的に分布していた一方,ほかの2つXl-TpS_harbおよびXl-TpS_Marは染色体Sのセットに特異的に分布していた.つまり,アフリカツメガエルの染色体Lと染色体Sのセットはサブゲノムに対応していることが明らかにされた.そこで,染色体Lと染色体Sに対応させてサブゲノムLとサブゲノムSと命名し,それらの起源となる絶滅した祖先種もLとSとした.また,それらのサブゲノムに属する同祖遺伝子は遺伝子名のあとに“.L”あるいは“.S”を付加して表記することにした.倍数体の動物種におけるサブゲノムの同定ははじめてのことであった.これにより,染色体Lと染色体Sに存在する遺伝子を調べることにより,異質倍数体化したのちのサブゲノムLとサブゲノムSの進化を解析することが可能になった.
3.サブゲノムLとサブゲノムSの進化
アフリカツメガエルにおいてサブゲノムのあいだの比較が可能になったことから,近縁種のネッタイツメガエルとアフリカツメガエルの2つのサブゲノムにおいて詳細な比較解析を行った.その結果,アフリカツメガエルの染色体のセットLはネッタイツメガエルの染色体とシンテニーや染色体の形態が非常に類似していたが,染色体のセットSは染色体のセットLと比べ染色体の長さが短く,染色体のレベルでの構造変化が多く生じていた.2倍体のネッタイツメガエルの遺伝子数は約21,000個であるが,アフリカツメガエルの遺伝子数はその約2倍の45,099個であった.これらのうち,嗅覚受容体遺伝子ファミリーのように多コピーで存在している遺伝子などを除いた24,419個の遺伝子のうち,異質倍数化により重複した2コピーが保存されている遺伝子は全体の56%である8806ペア(17,612遺伝子)であり,1コピーが欠失した遺伝子は6807遺伝子であった.そこで,染色体のセットごとの遺伝子の保存性について調べた結果,異質倍数化したのちサブゲノムLでは8.3%の遺伝子が消失していたのに対し,サブゲノムSでは31.5%の遺伝子が消失していた.
さらに,遺伝子の発現パターンや発現量についても比較した.14の異なる段階の卵母細胞あるいは発生段階の胚および14の異なる成体の組織を用いたRNA-Seq法により,大量のcDNAの配列情報を得た.これらをゲノム配列にマップして遺伝子の発現情報を得た.その結果,卵母細胞から母性-胚性転移期の胚にかけてはサブゲノムLの遺伝子の発現量がサブゲノムSの遺伝子より平均で12%高かったのに対し,母性-胚性転移期よりのちの胚および成体組織においてはサブゲノムLの遺伝子の発現量はサブゲノムSの遺伝子より25%高いことが明らかにされた.これらの結果から,アフリカツメガエルにおいては,1800万年前に祖先種Lと祖先種Sの種間交配およびゲノム重複による異質倍数化が起こり,そののち,サブゲノムLでは祖先種Lのゲノムをほぼそのまま残してきたのに対し,サブゲノムSでは多くの遺伝子の消失,偽遺伝子化,高頻度な染色体の再配列,そして,多くの遺伝子の発現量の低下が発生過程の胚および成体において起こったことが明らかにされた(図2).トウモロコシや出芽酵母の異質倍数体種においては,一方のサブゲノムの染色体領域の一部において多くの重複遺伝子の消失が起こっていると報告されている7,8).しかしながら,アフリカツメガエルはこれらとは異なり,サブゲノムSの全域において遺伝子の消失や染色体の再配列が生じていた.
おわりに
全ゲノム重複は生物の進化の過程においてしばしば起こる現象と考えられており,そのひとつの例が,約5億年前の古生代カンブリア紀に脊椎動物が出現する過程で起こったとされる2回の全ゲノム重複である9).全ゲノム重複により遺伝子の数を格段に増加させ,一部の重複遺伝子の機能の分担や新たな機能の獲得が脊椎動物の誕生とそののちの多様化および繁栄をもたらした要因であったと考えられている10).また,真骨魚類の祖先種において約3億年前に独自に全ゲノム重複が起こり,さらに,最近,全ゲノム配列が明らかにされたニジマスにおいては約1億年前にもう1回の全ゲノム重複が起こっている11).しかしながら,これらの全ゲノム重複からはいずれも1億年以上が経過しているため,祖先種に由来するサブゲノムを明らかにすることができず,サブゲノムごとの進化の過程を推測するのは困難であった.この研究においては,約1800万年前という比較的最近に全ゲノム重複の起こったアフリカツメガエルの全ゲノム配列を解読することにより,異質倍数体の動物種においてはじめてサブゲノムを識別することに成功し,それをもとに,全ゲノム重複ののちのサブゲノムの進化がはじめて明らかにされた.さらに,アフリカツメガエルと同じツメガエル属には,さらに1回あるいは2回の全ゲノム重複が想定される種が存在する12).これらの倍数体のツメガエルにおいてゲノムの構造を明らかにしアフリカツメガエルとの比較解析を行うことは,約5億年前に起こったとされる脊椎動物の共通祖先で生じた2回の全ゲノム重複や,そののち,真骨魚類に起こった全ゲノム重複が,のちの進化にどのようなインパクトをあたえたかを読み解く鍵,すなわち,ロゼッタストーンになるものと期待される.
この研究の成果はNature誌に掲載されただけでなく,このプロジェクトの日本チームにより8報のcompanion paperがDevelopmental Biology誌に掲載された13-20).
文 献
- Kobel, H. R. & Du Pasquier, L.: Genetics of polyploid Xenopus. Trends Genet., 2, 310-315 (1986)
- Hellsten, U., Harland, R. M., Gilchrist, M. J. et al.: The genome of the western clawed frog Xenopus tropicalis. Science, 328, 633-636 (2010)[PubMed]
- Kalhor, R., Tjong, H., Jayathilaka, N. et al.: Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol., 30, 90-98 (2012)[PubMed]
- Putnam, N. H., O’Connell, B. L., Stites, J. C. et al.: Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res., 26, 342-350 (2016)[PubMed]
- Matsuda, Y., Uno, Y., Kondo, M. et al.: A new nomenclature of Xenopus laevis chromosomes based on the phylogenetic relationship to Silurana/Xenopus tropicalis. Cytogenet. Genome Res., 145, 187-191 (2015)[PubMed]
- Uno, Y., Nishida, C., Takagi, C. et al.: Homoeologous chromosomes of Xenopus laevis are highly conserved after whole-genome duplication. Heredity, 111, 430-436 (2013)[PubMed]
- Schnable, J. C., Springer, N. M. & Freeling, M.: Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA, 108, 4069-4074 (2011)[PubMed]
- Marcet-Houben, M. & Gabaldon, T.: Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker’s yeast lineage. PLoS Biol., 13, e1002220 (2015)[PubMed]
- Holland, P. W., Garcia-Fernandez, J., Williams, N. A. et al.: Gene duplications and the origins of vertebrate development. Dev. Suppl., 1994, 125-133 (1994)[PubMed]
- Ohno, S.: Evolution by Gene Duplication. Springer, New York (1970)
- Berthelot, C., Brunet, F., Chalopin, D. et al.: The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun., 5, 3657 (2014)[PubMed]
- Evans, B. J., Carter, T. F., Greenbaum, E. et al.: Genetics, morphology, advertisement calls, and historical records distinguish six new polyploid species of African clawed frog (Xenopus, Pipidae) from west and central Africa. PLoS One, 10, e0142823 (2010)[PubMed]
- Suzuki, A., Uno, Y., Takahashi, S. et al.: Genome organization of the vg1 and nodal3 gene clusters in the allotetraploid frog Xenopus laevis. Dev. Biol., 426, 236-244 (2017)[PubMed]
- Haramoto, Y., Saijyo, T., Tanaka, T. et al.: Identification and comparative analyses of Siamois cluster genes in Xenopus laevis and tropicalis. Dev. Biol., 426, 374-383 (2017)[PubMed]
- Suzuki, A., Yoshida, H., van Heeringen, S. J. et al.: Genomic organization and modulation of gene expression of the TGF-β and FGF pathways in the allotetraploid frog Xenopus laevis. Dev. Biol., 426, 336-359 (2017)[PubMed]
- Watanabe, M., Yasuoka, Y., Mawaribuchi, S. et al.: Conservatism and variability of gene expression profiles among homeologous transcription factors in Xenopus laevis. Dev. Biol., 426, 301-324 (2017)[PubMed]
- Mawaribuchi, S., Takahashi, S., Wada, M. et al.: Sex chromosome differentiation and the W- and Z-specific loci in Xenopus laevis. Dev. Biol., 426, 393-400 (2017)[PubMed]
- Suzuki, K. T., Suzuki, M., Shigeta, M. et al.: Clustered Xenopus keratin genes: A genomic, transcriptomic, and proteomic analysis. Dev. Biol., 426, 384-392 (2017)[PubMed]
- Tanaka, T., Ochi, H., Takahashi, S. et al.: Genes coding for cyclin-dependent kinase inhibitors are fragile in Xenopus. Dev. Biol., 426, 291-300 (2017)[PubMed]
- Michiue, T., Yamamoto, T., Yasuoka, Y. et al.: High variability of expression profiles of homeologous genes for Wnt, Hh, Notch, and Hippo signaling pathways in Xenopus laevis. Dev. Biol., 426, 270-290 (2017)[PubMed]
著者プロフィール
略歴:2011年 北海道大学大学院生命科学院 修了,同年より名古屋大学大学院生命農学研究科 研究員.
研究テーマ:脊椎動物のゲノムおよび染色体の進化.
抱負:この研究におけるアフリカツメガエルの全ゲノム重複のように,ある生物種のゲノムや染色体を詳細に調べることにより,脊椎動物における進化の謎の一端を解明したい.
平良 眞規(Masanori Taira)
東京大学大学院理学系研究科 准教授.
研究室URL:http://www.biol.s.u-tokyo.ac.jp/users/lmb/lmb-hp.html
© 2016 宇野好宣・平良眞規 Licensed under CC 表示 2.1 日本
