クロマチンリモデリングタンパク質CHD8のハプロ不全は自閉症スペクトラムの原因となる
片山雄太・西山正章・中山敬一
(九州大学生体防御医学研究所 分子医科学分野)
email:中山敬一
DOI: 10.7875/first.author.2016.099
CHD8 haploinsufficiency results in autistic-like phenotypes in mice.
Yuta Katayama, Masaaki Nishiyama, Hirotaka Shoji, Yasuyuki Ohkawa, Atsuki Kawamura, Tetsuya Sato, Mikita Suyama, Toru Takumi, Tsuyoshi Miyakawa, Keiichi I. Nakayama
Nature, 537, 675-679 (2016)
自閉症スペクトラムは非常に発症頻度の高い発達障害であり,コミュニケーション能力の障害などにより社会活動に困難が生じる.自閉症スペクトラムの発症には遺伝的な要因が強く関与することから原因遺伝子がさかんに探索され,CHD8遺伝子がもっとも高頻度に変異していることが報告されたものの,これまで,CHD8の変異が自閉症スペクトラムの発症の原因であるという直接的な証拠はなかった.筆者らは,Chd8遺伝子をヘテロで欠損するマウスを作製し,CHD8と自閉症スペクトラムとの関連について検証した.その結果,CHD8ヘテロノックアウトマウスは自閉症スペクトラム様の行動異常を示したことから,CHD8のハプロ不全は自閉症スペクトラムの発症の原因であることが強く示唆された.さらに,CHD8ヘテロノックアウトマウスにおいては転写抑制因子であるRESTが異常に活性し,発生の初期から中期にかけての神経の発達が遅延していた.この結果から,CHD8のハプロ不全による神経の発達の遅延が自閉症スペクトラムの発症の原因であることが示唆された.
自閉症スペクトラムは社会性行動の障害および活動あるいは興味の範囲のいちじるしい限局性(固執傾向)をおもな特徴とする発達障害であり1),さらに,不安の増加や注意力の障害といったさまざまな症状が合併することが多い.発症の頻度は人口の1%以上と非常に高く,社会的な損失の大きさからも病態の解明および治療法の確立が急務となっている.自閉症スペクトラムの発症の原因には諸説あるものの,発生の初期から中期にかけての神経の発達の障害が関与するという説が有力視されている2).自閉症スペクトラムの発症には遺伝的な要因が強く関与することが知られており,これまでに多くの原因遺伝子の候補が報告されているが,とくに,シナプス関連タンパク質,Wnt-βカテニンシグナル伝達系,クロマチンリモデリングタンパク質にかかわる遺伝子の異常が自閉症スペクトラムの発症と強い相関を示す3).さらに近年,自閉症スペクトラムの患者を対象とした大規模なエキソーム解析により,CHD8遺伝子がもっとも頻度の高いde novo変異遺伝子として報告され,自閉症スペクトラムの原因遺伝子のもっとも有力な候補として注目をあつめている4-6).自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であり,これらの患者においては典型的な自閉症スペクトラムの症状にくわえ巨頭症および腸管の異常が多くみられる7).
CHD8はもともとWnt-βカテニンシグナル伝達系の抑制タンパク質として報告され当初はDuplinと命名されたが8),そののち,CHDファミリータンパク質の一員であることが明らかにされた.CHD8はクロマチンリモデリングタンパク質であり,クロマチンの構造を変化させることにより標的となる遺伝子の転写を制御する.筆者らは,これまでに,CHD8が転写因子であるp53やβカテニンと結合し,これらの転写制御領域にヒストンH1をリクルートすることによりその転写活性を抑制することを報告した9,10).Chd8遺伝子をホモで欠損したマウスは胎生の初期にp53の異常な活性化によるアポトーシスを起こし死亡することから,CHD8遺伝子は個体の発生において必須の役割を担うことがわかった11).
これらの報告から,CHD8のハプロ不全による遺伝子の発現制御の異常が自閉症スペクトラムの発症の原因であることが推測されたが,これまで,その発症機構はおろか,CHD8遺伝子の変異が自閉症スペクトラムの発症の原因であるという直接的な証拠も示されていなかった.
CHD8には全長の長鎖型CHD8とクロモドメインのみをもつ短鎖型CHD8が存在する(図1a).この2つのバリアントはN末端側を共通の領域としており,ともにp53とWnt-βカテニンシグナル伝達系の抑制能をもつ.一方,自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であり,CHD8遺伝子の全域に分布する(図1b).長鎖型CHD8に特異的な領域の変異は短鎖型CHD8の機能には影響しないと予想されたことから,とくに,長鎖型CHD8の機能の異常が自閉症スペクトラムの発症に強く関与すると推測された.これまでに筆者らは,長鎖型CHD8および短鎖型CHD8をともに欠損するマウスを作製し,このマウスはホモ欠損によりp53の異常な活性化によるアポトーシスを起こし胎生の初期に死亡することを報告した.今回,長鎖型CHD8を特異的に欠損するマウスを作製したところ,このマウスもホモ欠損により胎生致死となった.これらの結果から,長鎖型CHD8は短鎖型CHD8により代償されない特異的な機能をもつことがわかった.
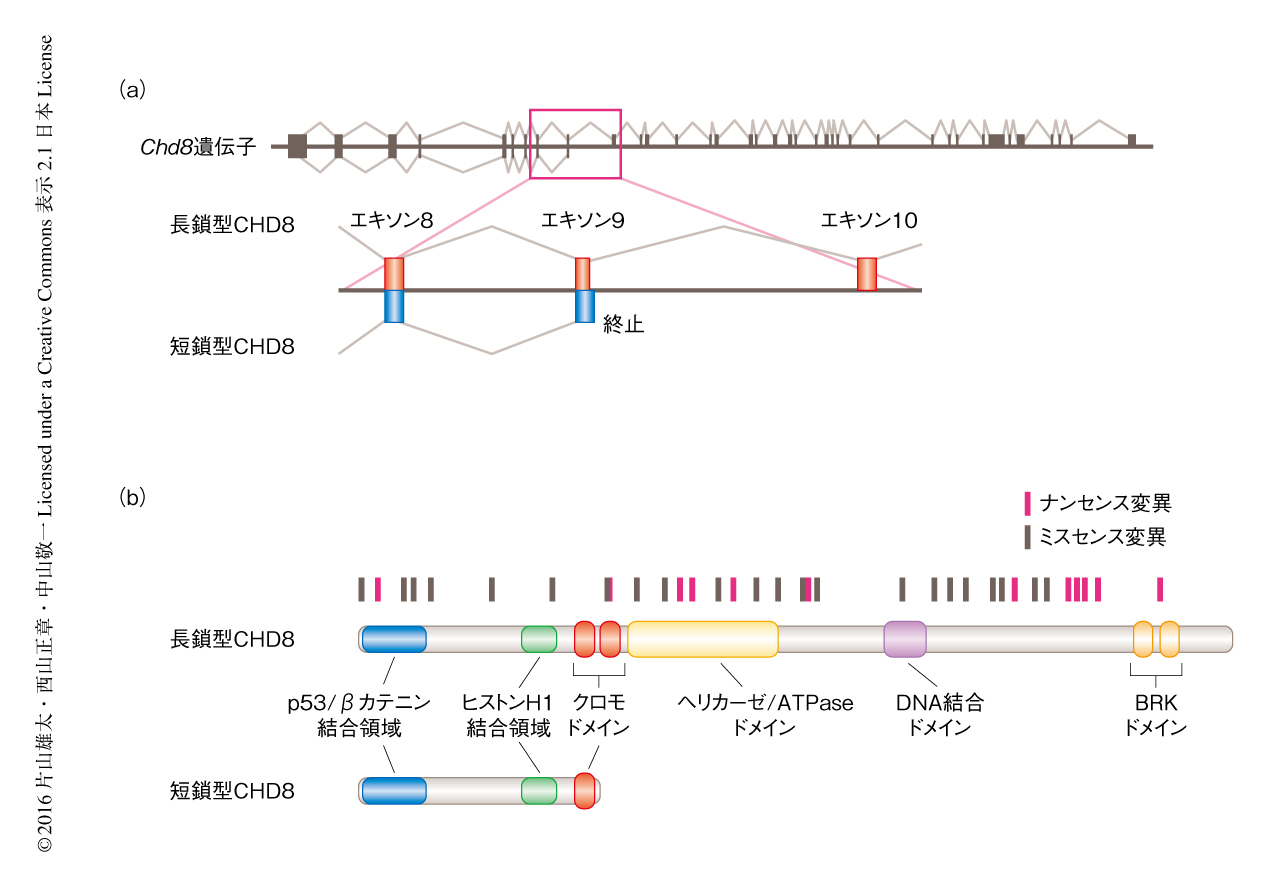
自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であったことから,CHD8のハプロ不全が自閉症スペクトラムの発症の原因であると推測し,長鎖型CHD8および短鎖型CHD8をともに欠損するマウス,および,長鎖型CHD8を特異的に欠損するマウスをそれぞれヘテロ変異として行動を解析した.すべての行動解析試験において,長鎖型CHD8および短鎖型CHD8をともに欠損するマウスと長鎖型CHD8を特異的に欠損するマウスは同じ表現型を示したため,この2つの系統を区別せず一括してCHD8ヘテロノックアウトマウスとする.CHD8ヘテロノックアウトマウスにおいて,Chd8遺伝子のmRNAの量は約50%に減少していたが,タンパク質の量は野生型のマウスの約70%であった.
CHD8遺伝子に変異をもつ自閉症スペクトラムの患者は巨頭症および腸管の異常を高頻度で合併する.CHD8ヘテロノックアウトマウスを観察したところ,成体のマウスの体重は変わらなかったものの,脳の重量が増加していた.腸管の運動を観察したところ,CHD8ヘテロノックアウトマウスは腸管の運動が低下する傾向を示した.この結果から,Chd8遺伝子のヘテロ欠損により自閉症スペクトラムの患者の特徴が再現されることが示された.
自閉症スペクトラムの患者においてよく観察される不安様の行動について,オープンフィールド試験,明暗選択箱試験,高架式十字迷路試験の3つの独立した行動試験により検証した.その結果,いずれの試験においてもCHD8ヘテロノックアウトマウスにおいては不安様の行動が増加したことから,この結果からも,自閉症スペクトラムの患者の特徴が再現されることが示された.
自閉症スペクトラムにおける主要な症状である固執傾向および社会性行動について検証した.記憶学習および固執傾向を評価するT字迷路試験により,CHD8ヘテロノックアウトマウスは記憶学習には異常がみられなかったものの,野生型のマウスと比較して強い固執傾向が観察された.社会性行動およびコミュニケーション能力を評価する社会的行動試験において,CHD8ヘテロノックアウトマウスは初対面のマウスの近くにいる時間は長くなったものの,マウスのコミュニケーションである追いかけ行動や臭い嗅ぎ行動は減少した.この行動異常は一部の自閉症スペクトラムの患者にみられる受動型とよばれるタイプのコミュニケーション障害に似ていた.
CHD8ヘテロノックアウトマウスにおいて観察されたこれらの表現型は,このマウスが自閉症スペクトラムを発症していることを示唆しており,CHD8のハプロ不全が自閉症スペクトラムの発症の原因となることを意味した.
CHD8はクロマチンリモデリングタンパク質であることから,CHD8の標的となる遺伝子の発現量の変化が自閉症スペクトラムの発症の原因であると推測された.CHD8の標的となる遺伝子を探索するため,マウスの脳の全体を試料としてChIP-seq法によりCHD8と結合するゲノム領域を同定した.CHD8の結合は66.9%が転写開始点の近傍に分布しており,これは全ゲノムに対する転写開始点の割合が約3%であることを考えると強い集積であった.そこで,CHD8は標的となる遺伝子の転写開始点と結合し,その転写量の制御にかかわると考えられた.CHD8の標的となる遺伝子の数は約9000と非常に多く,発現量の多い遺伝子の転写開始点ほどCHD8と強く結合していた.
野生型のマウスおよびCHD8ヘテロノックアウトマウスの脳の全体を試料としてRNA-seq法によりChd8遺伝子のヘテロ欠損により発現量の変化した遺伝子を探索した.しかし,予想に反して,顕著に発現量の変化した遺伝子はほとんどなかった.この結果,および,CHD8の標的となる遺伝子が非常に多いことをふまえ,少数の遺伝子の大きな変化ではなく,それぞれは小さい変動ながらも多数の遺伝子の変化の相乗効果が自閉症スペクトラムの発症に関与すると推測し,遺伝子の発現量の変化を個々の遺伝子ではなく遺伝子セットとして解析するGSEA(gene set enrichment analysis)法により再解析した.その結果,自閉症スペクトラムとの関連が報告されている遺伝子の発現がCHD8ヘテロノックアウトマウスにおいて低下していることが見い出された.さらに,自閉症スペクトラムの患者の脳において発現の低下している遺伝子も同様に,CHD8ヘテロノックアウトマウスにおける発現が低下していた.この結果から,CHD8ヘテロノックアウトマウスの脳における遺伝子の発現パターンは,自閉症スペクトラムの患者における遺伝子の発現パターンと類似していることが示された.発現の低下した遺伝子にはシナプス関連タンパク質やイオンチャネルといった神経の活性にかかわる遺伝子が多く含まれていたことから,CHD8ヘテロノックアウトマウスにおいて神経の機能の低下が自閉症スペクトラムの症状に関与する可能性が示唆された.
自閉症スペクトラムの発症には胎生の初期から中期にかけての神経の発達が関与すると考えられていることから,胎生14.5日齢のCHD8ヘテロノックアウトマウスの脳において遺伝子の発現量のデータを取得し,発生の初期あるいは中期においてそれぞれ高発現していた遺伝子セットについてGSEA法により解析した.その結果,胎生期のCHD8ヘテロノックアウトマウスの脳における遺伝子の発現パターンは,野生型のマウスと比較して,発生の初期に高発現する遺伝子の発現が優位であった.この結果から,CHD8ヘテロノックアウトマウスにおいては胎生の初期から中期の神経の発達が遅延していることが示唆された.
CHD8はWnt-βカテニンシグナル伝達系の抑制タンパク質であることから,CHD8のハプロ不全によるWnt-βカテニンシグナル伝達系の活性化が神経の発達の遅延の原因であると推測した.しかし,予想に反して,Wnt-βカテニンシグナル伝達系の活性化はほとんど認められず,非常に軽微な変化が胎生期のごくわずかな期間でのみ観察された.それに代わり,転写抑制因子RESTの標的となる遺伝子の発現がCHD8ヘテロノックアウトマウスにおいてもっとも顕著に低下していた.RESTは神経の分化の制御において重要であり,RESTの活性が低下することにより神経前駆細胞は神経細胞へと分化する.発生の中期において高発現していた遺伝子セットにもRESTの標的となる遺伝子が多く含まれていたことから,RESTの活性化により発生の中期にはたらく遺伝子の発現が抑制された結果,神経の発達に遅延が生じたと考えられた.さらに,RESTの標的となる遺伝子の発現の低下は自閉症スペクトラムの患者の死後脳のデータにもみられたことから,RESTの活性化はヒトの自閉症スペクトラムにも関与することが示唆された.
Chd8遺伝子のヘテロ欠損によりRESTが活性化したことから,CHD8はRESTの活性を抑制すると考えられた.胎生14.5日齢の野生型マウスの脳の抽出液を用いた共免疫沈降実験により,CHD8とRESTは物理的に結合することが明らかにされた.さらに,RESTの標的となる遺伝子をCHD8との結合の有無により2群に分け,それぞれにつきGSEA法により解析したところ,CHD8と結合し,かつ,RESTの標的となる遺伝子のみ,CHD8ヘテロノックアウトマウスにおいて発現の低下が観察された.この結果から,CHD8はRESTと結合しその標的となる遺伝子の発現を直接に制御すると考えられた.
神経の分化においてRESTはタンパク質分解および標的となるゲノム領域への結合の制御により活性が抑制されることが知られている.そこで,CHD8がこれらの抑制の機構を促進する可能性があると考えた.まず,野生型の細胞およびCHD8を欠損した細胞にタンパク質合成阻害剤であるシクロヘキシミドを処理しRESTの安定性を評価した.その結果,CHD8の欠損によりRESTの安定性に変化はみられず,CHD8はRESTの分解には関与しないことが示された.つづいて,野生型のマウスおよびCHD8ヘテロノックアウトマウスの脳を試料としてChIP-seq法によりRESTの結合量を調べたところ,RESTの標的となるゲノム領域への結合量に変化はなかった.これらの結果から,CHD8はRESTの分解あるいはゲノムへの結合のどちらにも関与しておらず,ヒストンの修飾や複合体の形成の阻害など,まったく別の機構によりRESTの活性を制御すると考えられた.
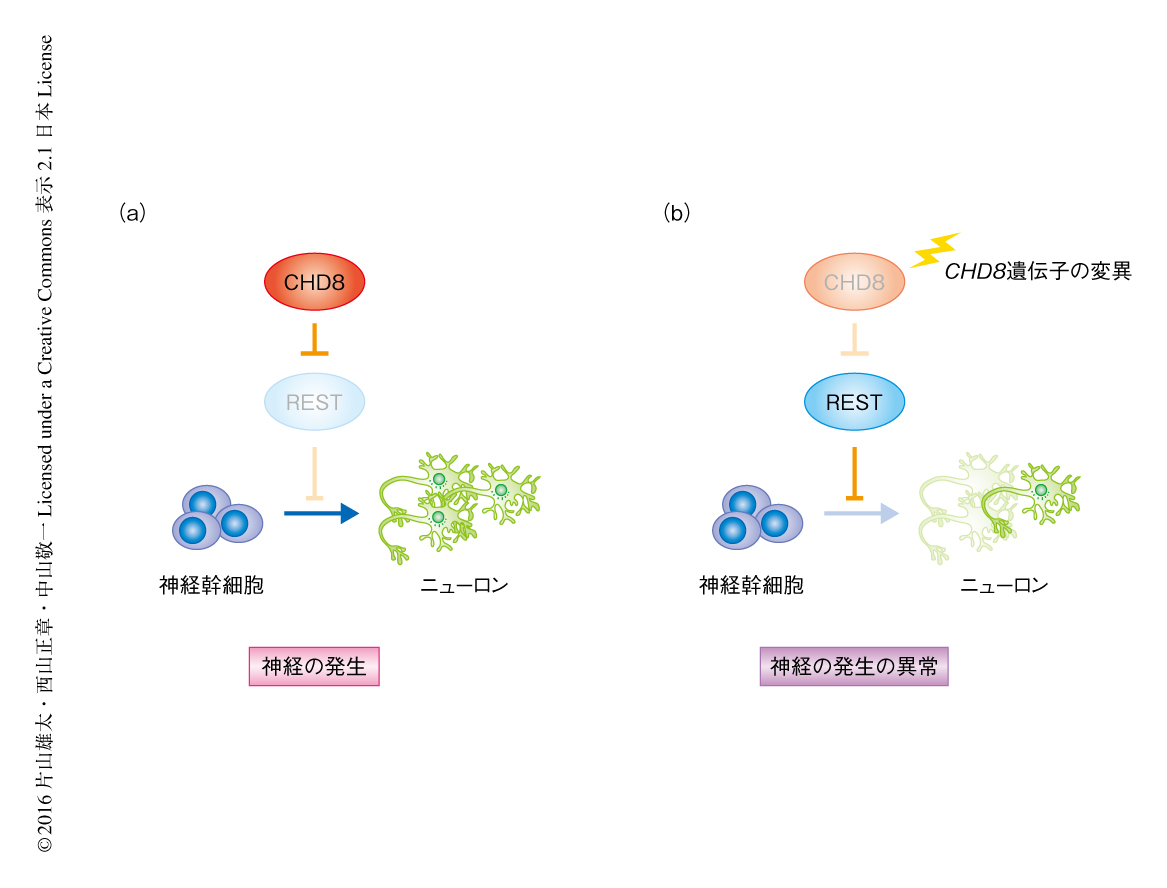
この研究は,自閉症スペクトラムの原因遺伝子の有力な候補として注目されているCHD8遺伝子の変異により自閉症スペクトラムが発症することを直接的に示した,はじめての報告である.さらに,CHD8遺伝子の変異はRESTの異常な活性化をともなう神経の発達の遅延をひき起こし,その結果,自閉症スペクトラムが発症するという仮説が提唱された(図2).CHD8はWnt-βカテニンシグナル伝達系の抑制タンパク質であるため,多くの研究者はWnt-βカテニンシグナル伝達系の活性化が自閉症スペクトラムの発症の原因であると予想していたが,CHD8ヘテロノックアウトマウスにおけるWnt-βカテニンシグナル伝達系の変化は軽微かつ一過性であり,おそらく,神経の発達に大きな影響をおよぼすレベルではないと思われた.一方で,CHD8とRESTの活性化との関連はこの研究における重要な発見のひとつであるが,いまのところ,CHD8によるRESTの抑制機構の詳細は明らかにされておらず,今後の課題となっている.
また,CHD8ヘテロノックアウトマウスは,原因遺伝子,遺伝子発現のパターン,行動学的な異常など,ヒトの自閉症スペクトラムの特徴を多方面から再現することにくわえ,独立した2つの系統のマウスにおいて同じ表現型が確認されていることから,非常に信頼性の高い自閉症スペクトラムのモデル動物であるといえよう.モデル動物は研究ツールとしてだけでなく薬剤スクリーニングにも大いに役だつことから,自閉症スペクトラムの治療法の開発にも貢献するものと考えている.さらに,自閉症スペクトラムとの関連が明らかにされたRESTは自閉症スペクトラムの新しい創薬ターゲットになりうる.すでに筆者らは,遺伝学的な手法およびREST阻害剤の投与による治療の実験を進めており,自閉症スペクトラムの治療法への応用について検討している.
略歴:九州大学生体防御医学研究所 学術研究員.
研究テーマ:クロマチンリモデリングタンパク質CHD8の遺伝子変異による自閉症スペクトラムの発症の機構.
西山 正章(Masaaki Nishiyama)
九州大学生体防御医学研究所 助教.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2016 片山雄太・西山正章・中山敬一 Licensed under CC 表示 2.1 日本
(九州大学生体防御医学研究所 分子医科学分野)
email:中山敬一
DOI: 10.7875/first.author.2016.099
CHD8 haploinsufficiency results in autistic-like phenotypes in mice.
Yuta Katayama, Masaaki Nishiyama, Hirotaka Shoji, Yasuyuki Ohkawa, Atsuki Kawamura, Tetsuya Sato, Mikita Suyama, Toru Takumi, Tsuyoshi Miyakawa, Keiichi I. Nakayama
Nature, 537, 675-679 (2016)
要 約
自閉症スペクトラムは非常に発症頻度の高い発達障害であり,コミュニケーション能力の障害などにより社会活動に困難が生じる.自閉症スペクトラムの発症には遺伝的な要因が強く関与することから原因遺伝子がさかんに探索され,CHD8遺伝子がもっとも高頻度に変異していることが報告されたものの,これまで,CHD8の変異が自閉症スペクトラムの発症の原因であるという直接的な証拠はなかった.筆者らは,Chd8遺伝子をヘテロで欠損するマウスを作製し,CHD8と自閉症スペクトラムとの関連について検証した.その結果,CHD8ヘテロノックアウトマウスは自閉症スペクトラム様の行動異常を示したことから,CHD8のハプロ不全は自閉症スペクトラムの発症の原因であることが強く示唆された.さらに,CHD8ヘテロノックアウトマウスにおいては転写抑制因子であるRESTが異常に活性し,発生の初期から中期にかけての神経の発達が遅延していた.この結果から,CHD8のハプロ不全による神経の発達の遅延が自閉症スペクトラムの発症の原因であることが示唆された.
はじめに
自閉症スペクトラムは社会性行動の障害および活動あるいは興味の範囲のいちじるしい限局性(固執傾向)をおもな特徴とする発達障害であり1),さらに,不安の増加や注意力の障害といったさまざまな症状が合併することが多い.発症の頻度は人口の1%以上と非常に高く,社会的な損失の大きさからも病態の解明および治療法の確立が急務となっている.自閉症スペクトラムの発症の原因には諸説あるものの,発生の初期から中期にかけての神経の発達の障害が関与するという説が有力視されている2).自閉症スペクトラムの発症には遺伝的な要因が強く関与することが知られており,これまでに多くの原因遺伝子の候補が報告されているが,とくに,シナプス関連タンパク質,Wnt-βカテニンシグナル伝達系,クロマチンリモデリングタンパク質にかかわる遺伝子の異常が自閉症スペクトラムの発症と強い相関を示す3).さらに近年,自閉症スペクトラムの患者を対象とした大規模なエキソーム解析により,CHD8遺伝子がもっとも頻度の高いde novo変異遺伝子として報告され,自閉症スペクトラムの原因遺伝子のもっとも有力な候補として注目をあつめている4-6).自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であり,これらの患者においては典型的な自閉症スペクトラムの症状にくわえ巨頭症および腸管の異常が多くみられる7).
CHD8はもともとWnt-βカテニンシグナル伝達系の抑制タンパク質として報告され当初はDuplinと命名されたが8),そののち,CHDファミリータンパク質の一員であることが明らかにされた.CHD8はクロマチンリモデリングタンパク質であり,クロマチンの構造を変化させることにより標的となる遺伝子の転写を制御する.筆者らは,これまでに,CHD8が転写因子であるp53やβカテニンと結合し,これらの転写制御領域にヒストンH1をリクルートすることによりその転写活性を抑制することを報告した9,10).Chd8遺伝子をホモで欠損したマウスは胎生の初期にp53の異常な活性化によるアポトーシスを起こし死亡することから,CHD8遺伝子は個体の発生において必須の役割を担うことがわかった11).
これらの報告から,CHD8のハプロ不全による遺伝子の発現制御の異常が自閉症スペクトラムの発症の原因であることが推測されたが,これまで,その発症機構はおろか,CHD8遺伝子の変異が自閉症スペクトラムの発症の原因であるという直接的な証拠も示されていなかった.
1.CHD8ヘテロノックアウトマウスの作製
CHD8には全長の長鎖型CHD8とクロモドメインのみをもつ短鎖型CHD8が存在する(図1a).この2つのバリアントはN末端側を共通の領域としており,ともにp53とWnt-βカテニンシグナル伝達系の抑制能をもつ.一方,自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であり,CHD8遺伝子の全域に分布する(図1b).長鎖型CHD8に特異的な領域の変異は短鎖型CHD8の機能には影響しないと予想されたことから,とくに,長鎖型CHD8の機能の異常が自閉症スペクトラムの発症に強く関与すると推測された.これまでに筆者らは,長鎖型CHD8および短鎖型CHD8をともに欠損するマウスを作製し,このマウスはホモ欠損によりp53の異常な活性化によるアポトーシスを起こし胎生の初期に死亡することを報告した.今回,長鎖型CHD8を特異的に欠損するマウスを作製したところ,このマウスもホモ欠損により胎生致死となった.これらの結果から,長鎖型CHD8は短鎖型CHD8により代償されない特異的な機能をもつことがわかった.
自閉症スペクトラムの患者において発見されたCHD8遺伝子の変異はすべてヘテロ変異であったことから,CHD8のハプロ不全が自閉症スペクトラムの発症の原因であると推測し,長鎖型CHD8および短鎖型CHD8をともに欠損するマウス,および,長鎖型CHD8を特異的に欠損するマウスをそれぞれヘテロ変異として行動を解析した.すべての行動解析試験において,長鎖型CHD8および短鎖型CHD8をともに欠損するマウスと長鎖型CHD8を特異的に欠損するマウスは同じ表現型を示したため,この2つの系統を区別せず一括してCHD8ヘテロノックアウトマウスとする.CHD8ヘテロノックアウトマウスにおいて,Chd8遺伝子のmRNAの量は約50%に減少していたが,タンパク質の量は野生型のマウスの約70%であった.
2.CHD8ヘテロノックアウトマウスは自閉症スペクトラム様の行動異常を示す
CHD8遺伝子に変異をもつ自閉症スペクトラムの患者は巨頭症および腸管の異常を高頻度で合併する.CHD8ヘテロノックアウトマウスを観察したところ,成体のマウスの体重は変わらなかったものの,脳の重量が増加していた.腸管の運動を観察したところ,CHD8ヘテロノックアウトマウスは腸管の運動が低下する傾向を示した.この結果から,Chd8遺伝子のヘテロ欠損により自閉症スペクトラムの患者の特徴が再現されることが示された.
自閉症スペクトラムの患者においてよく観察される不安様の行動について,オープンフィールド試験,明暗選択箱試験,高架式十字迷路試験の3つの独立した行動試験により検証した.その結果,いずれの試験においてもCHD8ヘテロノックアウトマウスにおいては不安様の行動が増加したことから,この結果からも,自閉症スペクトラムの患者の特徴が再現されることが示された.
自閉症スペクトラムにおける主要な症状である固執傾向および社会性行動について検証した.記憶学習および固執傾向を評価するT字迷路試験により,CHD8ヘテロノックアウトマウスは記憶学習には異常がみられなかったものの,野生型のマウスと比較して強い固執傾向が観察された.社会性行動およびコミュニケーション能力を評価する社会的行動試験において,CHD8ヘテロノックアウトマウスは初対面のマウスの近くにいる時間は長くなったものの,マウスのコミュニケーションである追いかけ行動や臭い嗅ぎ行動は減少した.この行動異常は一部の自閉症スペクトラムの患者にみられる受動型とよばれるタイプのコミュニケーション障害に似ていた.
CHD8ヘテロノックアウトマウスにおいて観察されたこれらの表現型は,このマウスが自閉症スペクトラムを発症していることを示唆しており,CHD8のハプロ不全が自閉症スペクトラムの発症の原因となることを意味した.
3.CHD8ヘテロノックアウトマウスは自閉症スペクトラムの患者における遺伝子の発現パターンを再現する
CHD8はクロマチンリモデリングタンパク質であることから,CHD8の標的となる遺伝子の発現量の変化が自閉症スペクトラムの発症の原因であると推測された.CHD8の標的となる遺伝子を探索するため,マウスの脳の全体を試料としてChIP-seq法によりCHD8と結合するゲノム領域を同定した.CHD8の結合は66.9%が転写開始点の近傍に分布しており,これは全ゲノムに対する転写開始点の割合が約3%であることを考えると強い集積であった.そこで,CHD8は標的となる遺伝子の転写開始点と結合し,その転写量の制御にかかわると考えられた.CHD8の標的となる遺伝子の数は約9000と非常に多く,発現量の多い遺伝子の転写開始点ほどCHD8と強く結合していた.
野生型のマウスおよびCHD8ヘテロノックアウトマウスの脳の全体を試料としてRNA-seq法によりChd8遺伝子のヘテロ欠損により発現量の変化した遺伝子を探索した.しかし,予想に反して,顕著に発現量の変化した遺伝子はほとんどなかった.この結果,および,CHD8の標的となる遺伝子が非常に多いことをふまえ,少数の遺伝子の大きな変化ではなく,それぞれは小さい変動ながらも多数の遺伝子の変化の相乗効果が自閉症スペクトラムの発症に関与すると推測し,遺伝子の発現量の変化を個々の遺伝子ではなく遺伝子セットとして解析するGSEA(gene set enrichment analysis)法により再解析した.その結果,自閉症スペクトラムとの関連が報告されている遺伝子の発現がCHD8ヘテロノックアウトマウスにおいて低下していることが見い出された.さらに,自閉症スペクトラムの患者の脳において発現の低下している遺伝子も同様に,CHD8ヘテロノックアウトマウスにおける発現が低下していた.この結果から,CHD8ヘテロノックアウトマウスの脳における遺伝子の発現パターンは,自閉症スペクトラムの患者における遺伝子の発現パターンと類似していることが示された.発現の低下した遺伝子にはシナプス関連タンパク質やイオンチャネルといった神経の活性にかかわる遺伝子が多く含まれていたことから,CHD8ヘテロノックアウトマウスにおいて神経の機能の低下が自閉症スペクトラムの症状に関与する可能性が示唆された.
4.転写抑制因子RESTの異常な活性化をともなう神経の発達の遅延
自閉症スペクトラムの発症には胎生の初期から中期にかけての神経の発達が関与すると考えられていることから,胎生14.5日齢のCHD8ヘテロノックアウトマウスの脳において遺伝子の発現量のデータを取得し,発生の初期あるいは中期においてそれぞれ高発現していた遺伝子セットについてGSEA法により解析した.その結果,胎生期のCHD8ヘテロノックアウトマウスの脳における遺伝子の発現パターンは,野生型のマウスと比較して,発生の初期に高発現する遺伝子の発現が優位であった.この結果から,CHD8ヘテロノックアウトマウスにおいては胎生の初期から中期の神経の発達が遅延していることが示唆された.
CHD8はWnt-βカテニンシグナル伝達系の抑制タンパク質であることから,CHD8のハプロ不全によるWnt-βカテニンシグナル伝達系の活性化が神経の発達の遅延の原因であると推測した.しかし,予想に反して,Wnt-βカテニンシグナル伝達系の活性化はほとんど認められず,非常に軽微な変化が胎生期のごくわずかな期間でのみ観察された.それに代わり,転写抑制因子RESTの標的となる遺伝子の発現がCHD8ヘテロノックアウトマウスにおいてもっとも顕著に低下していた.RESTは神経の分化の制御において重要であり,RESTの活性が低下することにより神経前駆細胞は神経細胞へと分化する.発生の中期において高発現していた遺伝子セットにもRESTの標的となる遺伝子が多く含まれていたことから,RESTの活性化により発生の中期にはたらく遺伝子の発現が抑制された結果,神経の発達に遅延が生じたと考えられた.さらに,RESTの標的となる遺伝子の発現の低下は自閉症スペクトラムの患者の死後脳のデータにもみられたことから,RESTの活性化はヒトの自閉症スペクトラムにも関与することが示唆された.
5.CHD8はRESTの活性を抑制する
Chd8遺伝子のヘテロ欠損によりRESTが活性化したことから,CHD8はRESTの活性を抑制すると考えられた.胎生14.5日齢の野生型マウスの脳の抽出液を用いた共免疫沈降実験により,CHD8とRESTは物理的に結合することが明らかにされた.さらに,RESTの標的となる遺伝子をCHD8との結合の有無により2群に分け,それぞれにつきGSEA法により解析したところ,CHD8と結合し,かつ,RESTの標的となる遺伝子のみ,CHD8ヘテロノックアウトマウスにおいて発現の低下が観察された.この結果から,CHD8はRESTと結合しその標的となる遺伝子の発現を直接に制御すると考えられた.
神経の分化においてRESTはタンパク質分解および標的となるゲノム領域への結合の制御により活性が抑制されることが知られている.そこで,CHD8がこれらの抑制の機構を促進する可能性があると考えた.まず,野生型の細胞およびCHD8を欠損した細胞にタンパク質合成阻害剤であるシクロヘキシミドを処理しRESTの安定性を評価した.その結果,CHD8の欠損によりRESTの安定性に変化はみられず,CHD8はRESTの分解には関与しないことが示された.つづいて,野生型のマウスおよびCHD8ヘテロノックアウトマウスの脳を試料としてChIP-seq法によりRESTの結合量を調べたところ,RESTの標的となるゲノム領域への結合量に変化はなかった.これらの結果から,CHD8はRESTの分解あるいはゲノムへの結合のどちらにも関与しておらず,ヒストンの修飾や複合体の形成の阻害など,まったく別の機構によりRESTの活性を制御すると考えられた.
おわりに
この研究は,自閉症スペクトラムの原因遺伝子の有力な候補として注目されているCHD8遺伝子の変異により自閉症スペクトラムが発症することを直接的に示した,はじめての報告である.さらに,CHD8遺伝子の変異はRESTの異常な活性化をともなう神経の発達の遅延をひき起こし,その結果,自閉症スペクトラムが発症するという仮説が提唱された(図2).CHD8はWnt-βカテニンシグナル伝達系の抑制タンパク質であるため,多くの研究者はWnt-βカテニンシグナル伝達系の活性化が自閉症スペクトラムの発症の原因であると予想していたが,CHD8ヘテロノックアウトマウスにおけるWnt-βカテニンシグナル伝達系の変化は軽微かつ一過性であり,おそらく,神経の発達に大きな影響をおよぼすレベルではないと思われた.一方で,CHD8とRESTの活性化との関連はこの研究における重要な発見のひとつであるが,いまのところ,CHD8によるRESTの抑制機構の詳細は明らかにされておらず,今後の課題となっている.
また,CHD8ヘテロノックアウトマウスは,原因遺伝子,遺伝子発現のパターン,行動学的な異常など,ヒトの自閉症スペクトラムの特徴を多方面から再現することにくわえ,独立した2つの系統のマウスにおいて同じ表現型が確認されていることから,非常に信頼性の高い自閉症スペクトラムのモデル動物であるといえよう.モデル動物は研究ツールとしてだけでなく薬剤スクリーニングにも大いに役だつことから,自閉症スペクトラムの治療法の開発にも貢献するものと考えている.さらに,自閉症スペクトラムとの関連が明らかにされたRESTは自閉症スペクトラムの新しい創薬ターゲットになりうる.すでに筆者らは,遺伝学的な手法およびREST阻害剤の投与による治療の実験を進めており,自閉症スペクトラムの治療法への応用について検討している.
文 献
- Abrahams, B. S. & Geschwind, D. H.: Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet., 9, 341-355 (2008)[PubMed]
- Willsey, A. J., Sanders, S. J., Li, M. et al.: Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 155, 997-1007 (2013)[PubMed]
- Krumm, N., O’Roak, B. J., Shendure, J. et al.: A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci., 37, 95-105 (2014)[PubMed]
- O’Roak, B. J., Vives, L., Girirajan, S. et al.: Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246-250 (2012)[PubMed]
- Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I. et al.: Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell, 149, 525-537 (2012)[PubMed]
- Neale, B. M., Kou, Y., Liu, L. et al.: Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242-245 (2012)[PubMed]
- Bernier, R., Golzio, C., Xiong, B. et al.: Disruptive CHD8 mutations define a subtype of autism early in development. Cell, 158, 263-276 (2014)[PubMed]
- Sakamoto, I., Kishida, S., Fukui, A. et al.: A novel β-catenin-binding protein inhibits β-catenin-dependent Tcf activation and axis formation. J. Biol. Chem., 275, 32871-32878 (2000)[PubMed]
- Nishiyama, M., Oshikawa, K., Tsukada, Y. et al.: CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol., 11, 172-182 (2009)[PubMed]
- Nishiyama, M., Skoultchi, A. I. & Nakayama, K. I.: Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol. Cell. Biol., 32, 501-512 (2012)[PubMed]
- Nishiyama, M., Nakayama, K., Tsunematsu, R. et al.: Early embryonic death in mice lacking the β-catenin-binding protein Duplin. Mol. Cell. Biol., 24, 8386-8394 (2004)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:九州大学生体防御医学研究所 学術研究員.
研究テーマ:クロマチンリモデリングタンパク質CHD8の遺伝子変異による自閉症スペクトラムの発症の機構.
西山 正章(Masaaki Nishiyama)
九州大学生体防御医学研究所 助教.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2016 片山雄太・西山正章・中山敬一 Licensed under CC 表示 2.1 日本
