mTOR複合体1とSIRT1は協調してカロリーの制限のもとでの腸幹細胞の増殖を制御する
五十嵐正樹・Leonard Guarente
(米国Massachusetts Institute of Technology,Department of Biology)
email:五十嵐正樹
DOI: 10.7875/first.author.2016.070
mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction.
Masaki Igarashi, Leonard Guarente
Cell, 166, 436-450 (2016)
寿命の延長にかかわるといわれるカロリーの制限はmTOR複合体1シグナルを低下させSIRT1の活性を上昇させると考えられている.筆者らは,この研究において,腸幹細胞におけるmTOR複合体1シグナルは,逆に,カロリーの制限により上昇することを明らかにした.カロリーの制限のもとでは,腸幹細胞のニッチを形成するPaneth細胞はcADPリボースを分泌し,隣接する腸幹細胞におけるSIRT1の活性の上昇およびS6K1の脱アセチル化によるリン酸化の促進をつうじmTOR複合体1シグナルを上昇させ,その結果,腸幹細胞におけるタンパク質の合成および腸幹細胞の数は増加した.mTORの阻害剤であるラパマイシンはカロリーの制限と同様な効果をもつと報告されているが,ラパマイシンはカロリーの制限における腸幹細胞の増加を抑制した.また,カロリーの制限にかかわる重要なシグナル伝達系を制御する薬剤は,異なる細胞において相反するはたらきをもつ可能性のあることが示唆された.
腸の組織は腸絨毛と腸陰窩からなり,腸陰窩は腸絨毛の底部に位置し,腸幹細胞とそのニッチ細胞であるPaneth細胞,および,腸絨毛の前駆細胞であるTA細胞を含む.腸の組織における恒常性は腸幹細胞の自己複製とTA細胞への分化との繊細なバランスにより保たれる.腸陰窩の底部にて腸幹細胞に隣接して存在するPaneth細胞は,腸幹細胞の維持に必要なWntシグナルなどの供給源となる.カロリーの制限のもとでは,Paneth細胞はmTOR複合体1の減少を介してcADPリボースの分泌を促進することにより腸幹細胞の自己複製を増加させることが示されているが1),腸幹細胞における増殖の制御機構については知られていなかった.そこで,筆者らは,カロリーの制限のもとでの腸幹細胞のPaneth細胞への応答の機構を明らかにすることを試みた.
NAD依存性アセチル化酵素であるSIRT1は,カロリーの制限における有益な効果の多くを媒介するといわれる2).腸に特異的なSIRT1ノックアウトマウスを作製し,25週間以上にわたりカロリーを制限した.野生型のマウスにおいてカロリーの制限により腸陰窩の深さは増加し腸絨毛の長さは減少したが,この効果は腸に特異的なSIRT1ノックアウトマウスにおいては認められなかった.また,カロリーの制限により細胞の増殖および腸幹細胞のマーカーに陽性を示す細胞の増加がみられたが,腸に特異的なSIRT1ノックアウトマウスにおいては認められなかった.これらの結果から,SIRT1はカロリーの制限における腸幹細胞の自己複製の増加および腸絨毛を形成する細胞への分化の低下において必要であることが示された.
SIRT1を過剰に発現するSIRT1トランスジェニックマウスを作製し解析したところ,カロリーを制限したマウスと同様に,腸陰窩の深さの増加,腸絨毛の長さの短縮,細胞の増殖,腸幹細胞のマーカーに陽性の細胞の増加が認められ,SIRT1の活性化はカロリーの制限における幹細胞の増加に必要かつ十分であることが示された.
腸幹細胞の増殖へのSIRT1の影響について詳細に調べるため,腸に特異的なSIRT1ノックアウトマウスおよびSIRT1を過剰に発現するトランスジェニックマウスから腸陰窩を単離し培養した.腸陰窩を培養すると腸幹細胞を起点として幹細胞およびそれから分化した細胞を含むコロニーが形成されるが,このコロニーの数は腸幹細胞の自己複製能を反映する.カロリーを制限したマウスから単離された腸陰窩は自由に摂食させたマウスから単離された腸陰窩と比べコロニーの数が増加したが,SIRT1ノックアウトマウスにおいてはカロリーの制限によりコロニーの数は増加しなかった.自由に摂食させたSIRT1を過剰発現するマウスから単離された腸陰窩は,カロリーを制限したマウスと同様に,コロニーの数が増加した.in vivoにおける結果と同様に,腸陰窩の培養のモデルにおいても,SIRT1の活性化がカロリーの制限おける幹細胞の増加に必要かつ十分であることが示された.
カロリーの制限のもとでのPaneth細胞による腸幹細胞の増加の促進においてSIRT1がどのように寄与するかを明らかにするため,同数の腸幹細胞とPaneth細胞とを共培養して形成されるコロニーの数を評価した.以前の報告1) と同様に,カロリーを制限したマウスに由来するPaneth細胞は,自由に摂食させたマウスに由来する腸幹細胞と共培養させるとコロニーの形成を促進した.しかし,この報告1) とは異なり,Paneth細胞の非存在下においては,カロリーを制限したマウスに由来する腸幹細胞は,自由に摂食させたマウスに由来する腸幹細胞より多くのコロニーを形成した.この相違は,腸陰窩を単離したときのEDTAの処理濃度および反応時間の違いによるものと考えられた.
SIRT1を過剰に発現するトランスジェニックマウスより腸幹細胞およびPaneth細胞を単離し共培養したところ,SIRT1を過剰発現するマウスに由来する腸幹細胞は,野生型マウスに由来する腸幹細胞に比べ多くのコロニーを形成した.しかし,SIRT1を過剰発現するマウスに由来するPaneth細胞は,野生型マウスに由来する腸幹細胞と共培養したときコロニーの形成を促進しなかった.
薬剤の添加により腸幹細胞において特異的にSIRT1を欠損するマウスを作製し,腸幹細胞を単離して,自由に摂食させたマウスあるいはカロリーを制限したマウスに由来するPaneth細胞と共培養した.SIRT1を欠損させた腸幹細胞はカロリーを制限したマウスに由来するPaneth細胞に反応しなかった.また,カロリーを制限した腸に特異的なSIRT1ノックアウトマウスに由来するPaneth細胞は,カロリーを制限した野生型マウスに由来するPaneth細胞と同様に,共培養において腸幹細胞からのコロニーの形成を促進した.これらの結果から,腸幹細胞におけるSIRT1の活性はカロリーの制限のもとでの腸幹細胞の自己複製の増加に必要かつ十分であることが示された.
カロリーの制限のもとでPaneth細胞が腸幹細胞の自己複製を促進するシグナル伝達系を探索した.Paneth細胞より分泌されるといわれるcADPリボースのアンタゴニストにより処理した腸幹細胞は,カロリーを制限したマウスに由来するPaneth細胞との共培養においてコロニーの数が増加しなかった.cADPリボースは細胞においてCa2+の濃度を上昇させることが報告されている3).Ca2+キレーターであるEGTAにより処理した腸幹細胞は,カロリーを制限したマウスに由来するPaneth細胞との共培養においてコロニーの数が増加しなかった.Ca2+はCa2+/カルモジュリン依存性キナーゼキナーゼを介してSIRT1を活性化することが報告されている4,5).腸幹細胞をCa2+/カルモジュリン依存性キナーゼキナーゼの特異的な阻害剤により処理したところ,カロリーを制限したマウスに由来するPaneth細胞との共培養におけるコロニーの数の増加は完全に抑制された.
Ca2+/カルモジュリン依存性キナーゼキナーゼはSIRT1をリン酸化することが知られているが4),カロリーの制限のもと腸陰窩においてSIRT1のリン酸化は増加せず,代わりに,腸陰窩および腸幹細胞においてCa2+/カルモジュリン依存性キナーゼキナーゼの別の基質であるAMPキナーゼのリン酸化が増加した.腸幹細胞をAMPキナーゼの特異的な阻害剤により処理したところ,カロリーを制限したマウスに由来するPaneth細胞との共培養におけるコロニーの数の増加は抑制された.
AMPキナーゼはNAD合成酵素であるNamptの発現の上昇およびNADの濃度の上昇を介し,SIRT1の活性を上昇させることが報告されている6,7).また,SIRT1はLKB1の脱アセチル化によりAMPキナーゼのリン酸化を促進する8,9).したがって,AMPキナーゼとSIRT1は正のフィードバックループを形成する.SIRT1を過剰に発現するトランスジェニックマウスに由来する腸陰窩においてAMPキナーゼのリン酸化の上昇,また,カロリーを制限したマウスに由来する腸陰窩においてLKB1のリン酸化の上昇が認められた.また,AMPキナーゼの活性化剤により処理した腸幹細胞においてコロニーの形成は促進された.さらに,カロリーを制限したマウスに由来する腸陰窩および腸幹細胞においてはNamptのmRNAレベルでの発現が上昇したが,AMPキナーゼの特異的な阻害剤により処理すると発現は低下した.以上から,腸幹細胞においてAMPキナーゼ,Nampt,SIRT1からなる正のフィードバックループの活性化が,Paneth細胞から分泌されたcADPリボースへの応答において中心的な役割をはたすことが示された.
SIRT1の標的となるシグナル伝達系の候補として,タンパク質の合成および細胞の増殖を規定するmTOR複合体1-S6K1シグナル伝達系に着目した.カロリーを制限したマウスに由来する腸陰窩および腸幹細胞において,S6K1のリン酸化およびS6のリン酸化の増加が認められた.mTOR複合体1に非依存性のS6K1のリン酸化およびmTOR複合体1の別の基質である4EBP1のリン酸化は,カロリーの制限により増加しなかった.また,カロリーの制限により腸絨毛におけるS6のリン酸化はかなり低下したが,腸幹細胞およびTA細胞におけるS6のリン酸化は逆に上昇することが免疫組織染色により見い出された.さらに,このS6のリン酸化の上昇およびS6K1のリン酸化は,腸幹細胞においてSIRT1を欠損させることにより完全に消失した.これらの結果から,SIRT1はmTOR複合体1-S6K1シグナル伝達系の活性化に必要であることが示された.また,AMPキナーゼを活性化させた腸陰窩およびSIRT1を過剰に発現するトランスジェニックマウスに由来する腸幹細胞において,S6のリン酸化およびS6K1のリン酸化は上昇した.
SIRT1によるS6K1の脱アセチル化によりS6K1のリン酸化が促進されることが報告されている10).カロリーを制限したマウスあるいはSIRT1を過剰に発現するトランスジェニックマウスに由来する腸陰窩においてS6K1のアセチル化は低下していた.また,腸に特異的なSIRT1ノックアウトマウスの腸陰窩においては,カロリーの制限下のもとでS6K1のアセチル化は低下しなかった.さらに,S6K1のC末端側にある3つのLysがSIRT1により脱アセチル化されることが報告されているが10),この3つのLysに変異を導入しアセチル化を欠失させたS6K1あるいはアセチル化を増加させたS6K1を腸幹細胞に発現させたところ,アセチル化の欠失したS6K1の発現は,カロリーを制限したマウスに由来するPaneth細胞との共培養と同様にコロニーの形成を促進したが,アセチル化の増加したS6K1の発現はコロニーの形成を抑制した.これらの結果から,カロリーを制限した腸幹細胞において,SIRT1がS6K1のアセチル化をつうじS6K1のリン酸化を促進することが示された.
S6K1のリン酸化がカロリーの制限のもとでの腸幹細胞の増加に寄与することをin vivoにおいて確かめるため,マウスにmTOR複合体1の阻害剤であるラパマイシンを処理したところ,自由に摂食させたマウスにおいては腸幹細胞のマーカーに陽性を示す細胞の数に影響はなかったが,カロリーを制限したマウスにおいて細胞の増殖および腸幹細胞のマーカーに陽性を示す細胞の増加が完全に抑制された.また,ラパマイシンの処理のもと25週間以上にわたりカロリーを制限したところ,カロリーの制限による腸陰窩の深さの増加および腸絨毛の長さの短縮は抑制された.
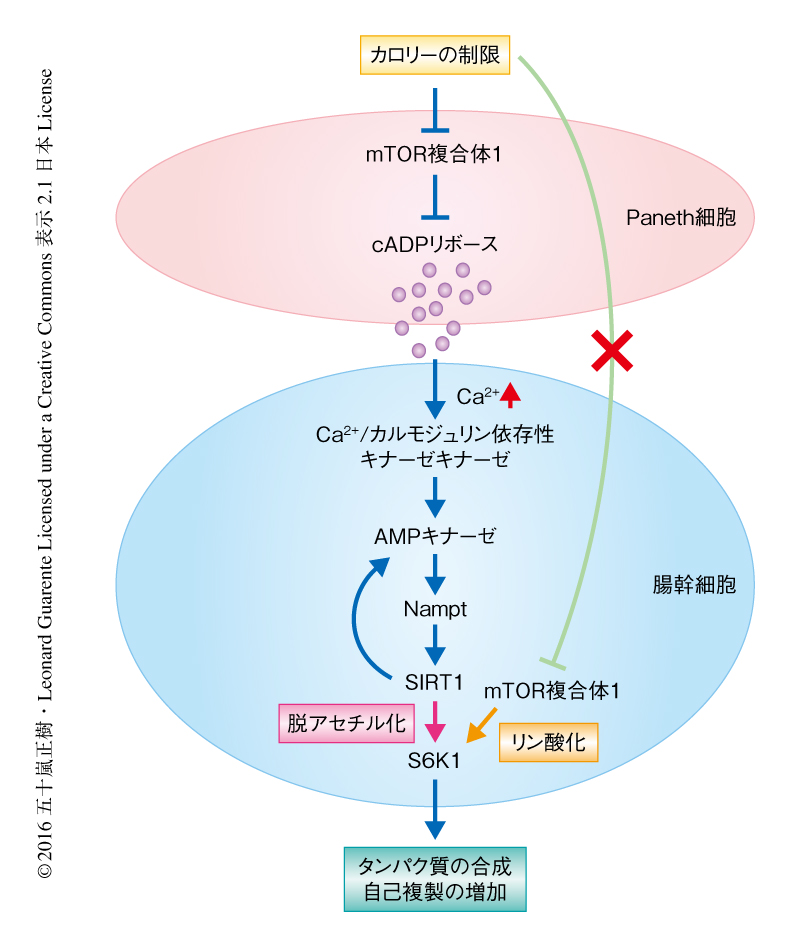
腸幹細胞およびそのニッチ細胞であるPaneth細胞は腸陰窩の底に隣接して配列しており,幹細胞とニッチ細胞の相互作用を調べるうえで腸は理想的な臓器といえる.Paneth細胞から分泌されるいくつかの物質が知られているが,ここでは,カロリーの制限下のもとで分泌されるcADPリボースに着目することにより,cADPリボースが腸幹細胞の増加を促進するシグナル伝達系について明らかにされた(図1).腸幹細胞におけるmTOR複合体1の応答は,Paneth細胞において報告されていた応答1) とは正反対であった.腸幹細胞においては,カロリーの制限のもとでのインスリンシグナルの低下やAMPキナーゼの活性化によるmTOR複合体1シグナルの低下などのエネルギーの感知機構は認められない.このような通常とは異なる状況におけるAMPキナーゼ,Nampt,SIRT1の活性化,SIRT1によるS6K1の脱アセチル化およびS6K1のリン酸化の上昇が,mTOR複合体1のカロリーの制限における正反対の応答を可能にした(図1).そして,カロリーの制限の効果を模倣するといわれるラパマイシンは,カロリーの制限における腸幹細胞の増殖を抑制した.この結果から,カロリーの制限の効果を模倣する薬剤を開発するうえでの困難さが示唆された.
略歴:2008年 東京大学大学院医学系研究科博士課程 修了,2010年より米国Massachusetts Institute of Technologyポストドクトラルフェロー.
研究テーマ:カロリーの制限および老化における幹細胞の制御.
関心事:老化におけるNADの制御.
Leonard Guarente
米国Massachusetts Institute of Technology教授.
研究室URL:http://web.mit.edu/biology/guarente/
© 2016 五十嵐正樹・Leonard Guarente Licensed under CC 表示 2.1 日本
(米国Massachusetts Institute of Technology,Department of Biology)
email:五十嵐正樹
DOI: 10.7875/first.author.2016.070
mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction.
Masaki Igarashi, Leonard Guarente
Cell, 166, 436-450 (2016)
要 約
寿命の延長にかかわるといわれるカロリーの制限はmTOR複合体1シグナルを低下させSIRT1の活性を上昇させると考えられている.筆者らは,この研究において,腸幹細胞におけるmTOR複合体1シグナルは,逆に,カロリーの制限により上昇することを明らかにした.カロリーの制限のもとでは,腸幹細胞のニッチを形成するPaneth細胞はcADPリボースを分泌し,隣接する腸幹細胞におけるSIRT1の活性の上昇およびS6K1の脱アセチル化によるリン酸化の促進をつうじmTOR複合体1シグナルを上昇させ,その結果,腸幹細胞におけるタンパク質の合成および腸幹細胞の数は増加した.mTORの阻害剤であるラパマイシンはカロリーの制限と同様な効果をもつと報告されているが,ラパマイシンはカロリーの制限における腸幹細胞の増加を抑制した.また,カロリーの制限にかかわる重要なシグナル伝達系を制御する薬剤は,異なる細胞において相反するはたらきをもつ可能性のあることが示唆された.
はじめに
腸の組織は腸絨毛と腸陰窩からなり,腸陰窩は腸絨毛の底部に位置し,腸幹細胞とそのニッチ細胞であるPaneth細胞,および,腸絨毛の前駆細胞であるTA細胞を含む.腸の組織における恒常性は腸幹細胞の自己複製とTA細胞への分化との繊細なバランスにより保たれる.腸陰窩の底部にて腸幹細胞に隣接して存在するPaneth細胞は,腸幹細胞の維持に必要なWntシグナルなどの供給源となる.カロリーの制限のもとでは,Paneth細胞はmTOR複合体1の減少を介してcADPリボースの分泌を促進することにより腸幹細胞の自己複製を増加させることが示されているが1),腸幹細胞における増殖の制御機構については知られていなかった.そこで,筆者らは,カロリーの制限のもとでの腸幹細胞のPaneth細胞への応答の機構を明らかにすることを試みた.
1.腸幹細胞においてSIRT1はカロリーの制限における腸幹細胞の増殖を媒介する
NAD依存性アセチル化酵素であるSIRT1は,カロリーの制限における有益な効果の多くを媒介するといわれる2).腸に特異的なSIRT1ノックアウトマウスを作製し,25週間以上にわたりカロリーを制限した.野生型のマウスにおいてカロリーの制限により腸陰窩の深さは増加し腸絨毛の長さは減少したが,この効果は腸に特異的なSIRT1ノックアウトマウスにおいては認められなかった.また,カロリーの制限により細胞の増殖および腸幹細胞のマーカーに陽性を示す細胞の増加がみられたが,腸に特異的なSIRT1ノックアウトマウスにおいては認められなかった.これらの結果から,SIRT1はカロリーの制限における腸幹細胞の自己複製の増加および腸絨毛を形成する細胞への分化の低下において必要であることが示された.
SIRT1を過剰に発現するSIRT1トランスジェニックマウスを作製し解析したところ,カロリーを制限したマウスと同様に,腸陰窩の深さの増加,腸絨毛の長さの短縮,細胞の増殖,腸幹細胞のマーカーに陽性の細胞の増加が認められ,SIRT1の活性化はカロリーの制限における幹細胞の増加に必要かつ十分であることが示された.
腸幹細胞の増殖へのSIRT1の影響について詳細に調べるため,腸に特異的なSIRT1ノックアウトマウスおよびSIRT1を過剰に発現するトランスジェニックマウスから腸陰窩を単離し培養した.腸陰窩を培養すると腸幹細胞を起点として幹細胞およびそれから分化した細胞を含むコロニーが形成されるが,このコロニーの数は腸幹細胞の自己複製能を反映する.カロリーを制限したマウスから単離された腸陰窩は自由に摂食させたマウスから単離された腸陰窩と比べコロニーの数が増加したが,SIRT1ノックアウトマウスにおいてはカロリーの制限によりコロニーの数は増加しなかった.自由に摂食させたSIRT1を過剰発現するマウスから単離された腸陰窩は,カロリーを制限したマウスと同様に,コロニーの数が増加した.in vivoにおける結果と同様に,腸陰窩の培養のモデルにおいても,SIRT1の活性化がカロリーの制限おける幹細胞の増加に必要かつ十分であることが示された.
カロリーの制限のもとでのPaneth細胞による腸幹細胞の増加の促進においてSIRT1がどのように寄与するかを明らかにするため,同数の腸幹細胞とPaneth細胞とを共培養して形成されるコロニーの数を評価した.以前の報告1) と同様に,カロリーを制限したマウスに由来するPaneth細胞は,自由に摂食させたマウスに由来する腸幹細胞と共培養させるとコロニーの形成を促進した.しかし,この報告1) とは異なり,Paneth細胞の非存在下においては,カロリーを制限したマウスに由来する腸幹細胞は,自由に摂食させたマウスに由来する腸幹細胞より多くのコロニーを形成した.この相違は,腸陰窩を単離したときのEDTAの処理濃度および反応時間の違いによるものと考えられた.
SIRT1を過剰に発現するトランスジェニックマウスより腸幹細胞およびPaneth細胞を単離し共培養したところ,SIRT1を過剰発現するマウスに由来する腸幹細胞は,野生型マウスに由来する腸幹細胞に比べ多くのコロニーを形成した.しかし,SIRT1を過剰発現するマウスに由来するPaneth細胞は,野生型マウスに由来する腸幹細胞と共培養したときコロニーの形成を促進しなかった.
薬剤の添加により腸幹細胞において特異的にSIRT1を欠損するマウスを作製し,腸幹細胞を単離して,自由に摂食させたマウスあるいはカロリーを制限したマウスに由来するPaneth細胞と共培養した.SIRT1を欠損させた腸幹細胞はカロリーを制限したマウスに由来するPaneth細胞に反応しなかった.また,カロリーを制限した腸に特異的なSIRT1ノックアウトマウスに由来するPaneth細胞は,カロリーを制限した野生型マウスに由来するPaneth細胞と同様に,共培養において腸幹細胞からのコロニーの形成を促進した.これらの結果から,腸幹細胞におけるSIRT1の活性はカロリーの制限のもとでの腸幹細胞の自己複製の増加に必要かつ十分であることが示された.
2.腸幹細胞におけるSIRT1の正のフィードバックループはカロリーの制限のもとでの腸幹細胞の増殖を制御する
カロリーの制限のもとでPaneth細胞が腸幹細胞の自己複製を促進するシグナル伝達系を探索した.Paneth細胞より分泌されるといわれるcADPリボースのアンタゴニストにより処理した腸幹細胞は,カロリーを制限したマウスに由来するPaneth細胞との共培養においてコロニーの数が増加しなかった.cADPリボースは細胞においてCa2+の濃度を上昇させることが報告されている3).Ca2+キレーターであるEGTAにより処理した腸幹細胞は,カロリーを制限したマウスに由来するPaneth細胞との共培養においてコロニーの数が増加しなかった.Ca2+はCa2+/カルモジュリン依存性キナーゼキナーゼを介してSIRT1を活性化することが報告されている4,5).腸幹細胞をCa2+/カルモジュリン依存性キナーゼキナーゼの特異的な阻害剤により処理したところ,カロリーを制限したマウスに由来するPaneth細胞との共培養におけるコロニーの数の増加は完全に抑制された.
Ca2+/カルモジュリン依存性キナーゼキナーゼはSIRT1をリン酸化することが知られているが4),カロリーの制限のもと腸陰窩においてSIRT1のリン酸化は増加せず,代わりに,腸陰窩および腸幹細胞においてCa2+/カルモジュリン依存性キナーゼキナーゼの別の基質であるAMPキナーゼのリン酸化が増加した.腸幹細胞をAMPキナーゼの特異的な阻害剤により処理したところ,カロリーを制限したマウスに由来するPaneth細胞との共培養におけるコロニーの数の増加は抑制された.
AMPキナーゼはNAD合成酵素であるNamptの発現の上昇およびNADの濃度の上昇を介し,SIRT1の活性を上昇させることが報告されている6,7).また,SIRT1はLKB1の脱アセチル化によりAMPキナーゼのリン酸化を促進する8,9).したがって,AMPキナーゼとSIRT1は正のフィードバックループを形成する.SIRT1を過剰に発現するトランスジェニックマウスに由来する腸陰窩においてAMPキナーゼのリン酸化の上昇,また,カロリーを制限したマウスに由来する腸陰窩においてLKB1のリン酸化の上昇が認められた.また,AMPキナーゼの活性化剤により処理した腸幹細胞においてコロニーの形成は促進された.さらに,カロリーを制限したマウスに由来する腸陰窩および腸幹細胞においてはNamptのmRNAレベルでの発現が上昇したが,AMPキナーゼの特異的な阻害剤により処理すると発現は低下した.以上から,腸幹細胞においてAMPキナーゼ,Nampt,SIRT1からなる正のフィードバックループの活性化が,Paneth細胞から分泌されたcADPリボースへの応答において中心的な役割をはたすことが示された.
3.カロリーの制限のもとSIRT1は腸幹細胞におけるS6K1のリン酸化をS6K1の脱アセチル化をとおし促進する
SIRT1の標的となるシグナル伝達系の候補として,タンパク質の合成および細胞の増殖を規定するmTOR複合体1-S6K1シグナル伝達系に着目した.カロリーを制限したマウスに由来する腸陰窩および腸幹細胞において,S6K1のリン酸化およびS6のリン酸化の増加が認められた.mTOR複合体1に非依存性のS6K1のリン酸化およびmTOR複合体1の別の基質である4EBP1のリン酸化は,カロリーの制限により増加しなかった.また,カロリーの制限により腸絨毛におけるS6のリン酸化はかなり低下したが,腸幹細胞およびTA細胞におけるS6のリン酸化は逆に上昇することが免疫組織染色により見い出された.さらに,このS6のリン酸化の上昇およびS6K1のリン酸化は,腸幹細胞においてSIRT1を欠損させることにより完全に消失した.これらの結果から,SIRT1はmTOR複合体1-S6K1シグナル伝達系の活性化に必要であることが示された.また,AMPキナーゼを活性化させた腸陰窩およびSIRT1を過剰に発現するトランスジェニックマウスに由来する腸幹細胞において,S6のリン酸化およびS6K1のリン酸化は上昇した.
SIRT1によるS6K1の脱アセチル化によりS6K1のリン酸化が促進されることが報告されている10).カロリーを制限したマウスあるいはSIRT1を過剰に発現するトランスジェニックマウスに由来する腸陰窩においてS6K1のアセチル化は低下していた.また,腸に特異的なSIRT1ノックアウトマウスの腸陰窩においては,カロリーの制限下のもとでS6K1のアセチル化は低下しなかった.さらに,S6K1のC末端側にある3つのLysがSIRT1により脱アセチル化されることが報告されているが10),この3つのLysに変異を導入しアセチル化を欠失させたS6K1あるいはアセチル化を増加させたS6K1を腸幹細胞に発現させたところ,アセチル化の欠失したS6K1の発現は,カロリーを制限したマウスに由来するPaneth細胞との共培養と同様にコロニーの形成を促進したが,アセチル化の増加したS6K1の発現はコロニーの形成を抑制した.これらの結果から,カロリーを制限した腸幹細胞において,SIRT1がS6K1のアセチル化をつうじS6K1のリン酸化を促進することが示された.
4.mTOR複合体1の阻害剤はカロリーの制限のもとでの腸幹細胞の増殖を抑制する
S6K1のリン酸化がカロリーの制限のもとでの腸幹細胞の増加に寄与することをin vivoにおいて確かめるため,マウスにmTOR複合体1の阻害剤であるラパマイシンを処理したところ,自由に摂食させたマウスにおいては腸幹細胞のマーカーに陽性を示す細胞の数に影響はなかったが,カロリーを制限したマウスにおいて細胞の増殖および腸幹細胞のマーカーに陽性を示す細胞の増加が完全に抑制された.また,ラパマイシンの処理のもと25週間以上にわたりカロリーを制限したところ,カロリーの制限による腸陰窩の深さの増加および腸絨毛の長さの短縮は抑制された.
おわりに
腸幹細胞およびそのニッチ細胞であるPaneth細胞は腸陰窩の底に隣接して配列しており,幹細胞とニッチ細胞の相互作用を調べるうえで腸は理想的な臓器といえる.Paneth細胞から分泌されるいくつかの物質が知られているが,ここでは,カロリーの制限下のもとで分泌されるcADPリボースに着目することにより,cADPリボースが腸幹細胞の増加を促進するシグナル伝達系について明らかにされた(図1).腸幹細胞におけるmTOR複合体1の応答は,Paneth細胞において報告されていた応答1) とは正反対であった.腸幹細胞においては,カロリーの制限のもとでのインスリンシグナルの低下やAMPキナーゼの活性化によるmTOR複合体1シグナルの低下などのエネルギーの感知機構は認められない.このような通常とは異なる状況におけるAMPキナーゼ,Nampt,SIRT1の活性化,SIRT1によるS6K1の脱アセチル化およびS6K1のリン酸化の上昇が,mTOR複合体1のカロリーの制限における正反対の応答を可能にした(図1).そして,カロリーの制限の効果を模倣するといわれるラパマイシンは,カロリーの制限における腸幹細胞の増殖を抑制した.この結果から,カロリーの制限の効果を模倣する薬剤を開発するうえでの困難さが示唆された.
文 献
- Yilmaz, O. H., Katajisto, P., Lamming, D. W. et al.: mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature, 486, 490-495 (2012)[PubMed]
- Guarente, L.: Calorie restriction and sirtuins revisited. Genes Dev., 27, 2072-2085 (2013)[PubMed]
- De Flora, A., Zocchi, E., Guida, L. et al.: Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. NY Acad. Sci., 1028, 176-191 (2004)[PubMed]
- Wen, L., Chen, Z., Zhang, F. et al.: Ca2+/calmodulin-dependent protein kinase kinase β phosphorylation of Sirtuin 1 in endothelium is athero-protective. Proc. Natl. Acad. Sci. USA, 110, E2420-E2427 (2013)[PubMed]
- Iwabu, M., Yamauchi, T., Okada-Iwabu, M. et al.: Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature, 464, 1313-1319 (2010)[PubMed]
- Brandauer, J., Vienberg, S. G., Andersen, M. A. et al.: AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J. Physiol., 591, 5207-5220 (2013)[PubMed]
- Canto, C., Gerhart-Hines, Z., Feige, J. N. et al.: AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature, 458, 1056-1060 (2009)[PubMed]
- Hou, X., Xu, S., Maitland-Toolan, K. A. et al.: SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem., 283, 20015-20026 (2008)[PubMed]
- Lan, F., Cacicedo, J. M., Ruderman, N. et al.: SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem., 283, 27628-27635.(2008)[PubMed]
- Hong, S., Zhao, B., Lombard, D. B. et al.: Crosstalk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J. Biol. Chem., 289, 13132-13141 (2014)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2008年 東京大学大学院医学系研究科博士課程 修了,2010年より米国Massachusetts Institute of Technologyポストドクトラルフェロー.
研究テーマ:カロリーの制限および老化における幹細胞の制御.
関心事:老化におけるNADの制御.
Leonard Guarente
米国Massachusetts Institute of Technology教授.
研究室URL:http://web.mit.edu/biology/guarente/
© 2016 五十嵐正樹・Leonard Guarente Licensed under CC 表示 2.1 日本
