アディポネクチンは骨髄において炎症性サイトカインの産生を抑制することにより造血前駆細胞の細菌の感染に対する応答を高める
正本庸介・黒川峰夫
(東京大学大学院医学系研究科 血液・腫瘍病態学講座)
email:正本庸介,黒川峰夫
DOI: 10.7875/first.author.2016.069
Adiponectin enhances antibacterial activity of hematopoietic cells by suppressing bone marrow inflammation.
Yosuke Masamoto, Shunya Arai, Tomohiko Sato, Akihide Yoshimi, Naoto Kubota, Iseki Takamoto, Yoichiro Iwakura, Akihiko Yoshimura, Takashi Kadowaki, Mineo Kurokawa
Immunity, 44, 1422-1433 (2016)
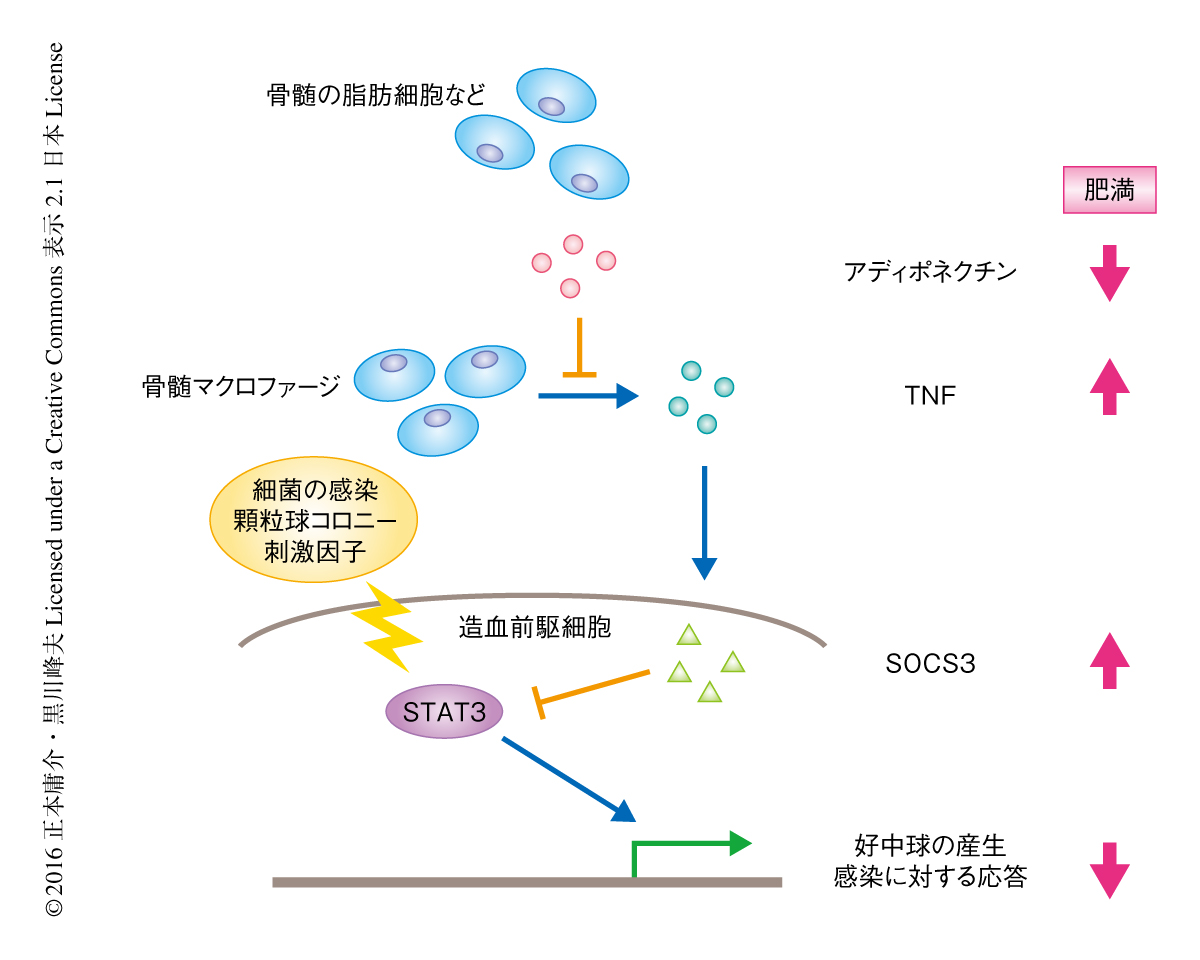
肥満症によりさまざまな感染症の頻度および重症度の上がることが知られているが,その分子機序には不明な点が多い.今回,筆者らは,肥満モデルマウスの骨髄において,脂肪細胞の産生する“善玉”ホルモンであるアディポネクチンの濃度が低下し,それにともない炎症性サイトカインであるTNFの骨髄マクロファージからの分泌が慢性的に亢進することを示した.炎症性サイトカインに慢性的に曝露されることにより,骨髄の造血前駆細胞においてサイトカインシグナル抑制タンパク質であるSOCS3の発現が上昇し,細菌に感染した際のSTAT3シグナル伝達系の活性化が阻害される結果,感染からの防御に必須な顆粒球が骨髄において十分に産生されず細菌の駆逐が抑制された.肥満マウスにアディポネクチンあるいはアディポネクチン受容体のアゴニストであるAdipoRonを投与することにより,細菌に感染した際の顆粒球の産生は回復し重症化が抑制された.この結果から,アディポネクチンは肥満に関連した感染症の治療において標的になりうると考えられた.
肥満症は心血管疾患,悪性腫瘍,感染症などさまざまな合併症と関連することが知られ,肥満症においてはさまざまな感染症の頻度および重症度が上がることが臨床的な研究により示唆されているが1),その分子機序には不明な点が多い.肥満症における中心的な病態は,脂肪細胞から分泌されるサイトカインであるアディポカインの,脂肪細胞の機能の異常にともなう産生の異常であると考えられている.肥満症においては脂肪細胞が肥大化しTNFやインターロイキン6などの炎症性サイトカインの産生が亢進する一方,アディポネクチンなど抗炎症作用をもつアディポカインの産生は逆説的に低下し,肥満症および合併症の病態を形成する.造血の主座である骨髄には脂肪細胞が豊富に存在し,肥満症における脂肪細胞の機能の異常は造血にもなんらかの影響をおよぼすと考えられるが,そのような検討はこれまでほとんどなされていない.今回,筆者らは,肥満症と感染症とのかかわりに注目し,肥満症における造血の異常が感染の重症化につながるという仮説をたてこれをマウスにおいて検証するとともに,肥満症における感染の重症化に対する治療的なアプローチについて検討した.
細菌の感染に対する適切な応答には,細菌を貪食する主体となる好中球が十分に産生されることが必要である.感染にともない内因性に産生されるサイトカインである顆粒球コロニー刺激因子は,骨髄における顆粒球前駆細胞の増殖および骨髄からの遊走を強く促進し,細菌の感染に対する応答にもっとも重要な役割をはたしており,顆粒球コロニー刺激因子を投与すると細菌に感染したときと類似した応答をひき起こすことが知られている2).汎用される肥満モデルマウスのひとつである高脂肪食を負荷したマウスに顆粒球コロニー刺激因子を投与すると,対照となるマウスにおいてみられた骨髄における顆粒球前駆細胞の十分な増殖がみられず,好中球の産生が低下した.
肥満モデルマウスの骨髄においてさまざまなサイトカインの発現を調べた研究において多くのサイトカインの発現量は変化しないことが報告されていたため3),多彩な生理的な機能をもつにもかかわらず,これまで,in vivoにおいて造血に対する作用が調べられていなかったアディポネクチンに注目した.肥満マウスにてアディポネクチンの血清における濃度は保たれていたが,骨髄における濃度は低下しており,骨髄においてアディポネクチンは血清とは異なる機構により制御されることが示唆された.
アディポネクチンノックアウトマウスは耐糖能の異常,動脈硬化への感受性の上昇などメタボリック症候群に類似した表現型を示すが,それらはいずれも軽微であることが知られている4).アディポネクチンノックアウトマウスの造血系には,細胞の数,分化,アポトーシス,細胞周期,造血幹細胞あるいは前駆細胞の活性を示す細胞の頻度など,さまざまな指標において明らかな異常はみられなかった.しかし,アディポネクチンノックアウトマウスに顆粒球コロニー刺激因子を投与したところ,肥満マウスと同様に,顆粒球前駆細胞の増殖の低下および好中球の産生の低下が認められた.
リステリア菌は細胞内寄生性のグラム陽性桿菌であり,おもに免疫不全者に対し重篤な全身感染症を起こす.リステリア菌の排除には顆粒球コロニー刺激因子によるシグナルの機能することが必要であることが示されており,リステリア菌の腹腔への感染はマウスにおける細菌感染モデルとして汎用される.この細菌感染モデルを用いて,肥満マウスおよびアディポネクチンノックアウトマウスの細菌の感染に対する応答を観察した.これらのマウスにおいては感染ののち骨髄および末梢血における好中球の増加,および,感染した部位への好中球の遊走が障害され,感染ののち7日目の肝臓および脾臓には有意に多数の細菌が残存しており感染は重症化していると考えられた.組換え型アディポネクチンの静脈内投与,あるいは,アディポネクチン受容体の小分子アゴニストであるAdipoRonの経口投与により,これらの感染に対する応答はいずれも回復し,細菌の数は低下した.
顆粒球コロニー刺激因子による顆粒球前駆細胞の増殖においては,STAT3シグナル伝達系の活性化が中心的な役割をはたすことが知られている.肥満マウスおよびアディポネクチンノックアウトマウスの造血細胞をin vitroにおいて顆粒球コロニー刺激因子により刺激したところ,対照となるマウスと比較して,STAT3のリン酸化が低下していた.そこで,これらのマウスにin vivoにおいて顆粒球コロニー刺激因子を投与したのち造血細胞を回収すると,STAT3のリン酸化が低下し,STAT3の下流の主要なエフェクターとして知られるC/EBPβの発現5) が低下していた.同様に,肥満マウスおよびアディポネクチンノックアウトマウスにおいて細菌の感染によりSTAT3シグナル伝達系の活性の低下がみられた.いずれのマウスにおいても,in vivoにおいてアディポネクチンを投与することによりSTAT3シグナル伝達系の活性化は回復した.
肥満マウスおよびアディポネクチンノックアウトマウスの造血細胞における顆粒球コロニー刺激因子に対する応答の低下の機序を明らかにするため,顆粒球コロニー刺激因子に応答した増殖の主体になると考えられる造血前駆細胞について詳細に解析した6).その結果,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化は,多能性前駆細胞,骨髄系共通前駆細胞,顆粒球マクロファージ前駆細胞といった前駆細胞の画分において顕著に低下していた.肥満モデルマウスの肝臓および筋肉においてSTAT3シグナル伝達系の主要な抑制タンパク質であるSOCS3の発現が上昇しインスリンのシグナルを抑制することが知られていたことから7),造血系においてSOCS3の発現を比較したところ,肥満マウスおよびアディポネクチンノックアウトマウスの造血前駆細胞においてSOCS3の発現が上昇し,in vivoにおけるアディポネクチンの投与によりこの発現は低下した.肥満マウスにおいて造血細胞の全体にてSOCS3の発現を低下させると,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化および細菌の感染に対する応答が回復した.一方で,肥満マウスにおいて成熟した顆粒球およびマクロファージにてSOCS3の発現を低下させても,これらの表現型に変化はみられなかった.以上のことから,アディポネクチンの欠乏した状態では造血前駆細胞におけるSOCS3の発現が上昇し,それによりSTAT3シグナル伝達系の活性化が抑制されると考えられた.
造血前駆細胞をin vitroにおいてアディポネクチンにより処理してもSOCS3の発現量に変化はみられなかった.そこで,生体においては,アディポネクチンにより発現が抑制されSOCS3の発現を誘導するタンパク質が作用するのではないかと考え,候補として,炎症性サイトカインであるTNFに注目した8-10).実際に,in vitroにおいてTNFは造血前駆細胞におけるSOCS3の発現を誘導し,また,肥満マウスおよびアディポネクチンノックアウトマウスの骨髄においてTNFの濃度が上昇しており,アディポネクチンの投与によりTNFの濃度は低下した.TNFノックアウトマウスにおいては肥満により造血前駆細胞におけるSOCS3の発現は上昇せず,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化が回復したことから,TNFが重要なはたらきをすることが示唆された.
骨髄は血清と比較してTNFの濃度が高いことから,骨髄においては局所的にTNFが産生されると考えられた.骨髄のさまざまな細胞において比較したところ,多くの造血細胞の画分においてTNFの産生がみられたが,アディポネクチンによるTNFの産生の抑制はF4/80陽性のマクロファージでのみ観察された.そこで,薬理学的にマクロファージを消失させin vivoにおいて顆粒球コロニー刺激因子に対する応答を比較したところ,アディポネクチンノックアウトマウスにおいて細菌に感染した際の顆粒球の産生の低下はみられなくなり,アディポネクチンの作用はマクロファージを介することが示唆された.
アディポネクチンが骨髄マクロファージにおいてTNFの産生を抑制する機構について検討した.さまざまな細胞種を用いた実験においてアディポネクチンはリポ多糖により刺激したときのTNFの発現を転写レベルにおいて直接的に抑制することが知られているが11),生体のように,リポ多糖のようなTNFを産生させる強力な刺激が存在せず,長時間にわたりアディポネクチンに曝露される状態において,アディポネクチンがTNFの産生にどのように影響するかは不明であった.そこで,アディポネクチンノックアウトマウスの骨髄からマクロファージを単離し,リポ多糖の添加あるいは非添加の条件においてアディポネクチンを処理したところ,アディポネクチンはリポ多糖により刺激したときのTNFの発現を転写レベルで強力に抑制したものの,リポ多糖の刺激のないときのTNFの発現に対する抑制の効果は比較的小さかった.そのため,アディポネクチンが骨髄マクロファージに直接的に作用してTNFの産生を抑制する以外にも,なんらかの機序の存在が示唆された.
おもにin vitroにおける実験において,マクロファージは周囲のサイトカインに応答して炎症性のM1マクロファージあるいは抗炎症性のM2マクロファージに分化することが知られている.そこで,肥満マウスおよびアディポネクチンノックアウトマウスから骨髄マクロファージを単離して遺伝子の発現を比較したところ,TNF遺伝子を含むM1マクロファージと関連する複数の遺伝子の発現の上昇がみられ,また,M1マクロファージの形質と関連することの知られているMHCクラスII分子を発現するマクロファージが増加した.肥満マウス,アディポネクチンノックアウトマウス,対照となるマウスのいずれにおいても,MHCクラスII分子を発現する骨髄マクロファージはTNFを多く産生しており,MHCクラスII分子を発現する骨髄マクロファージのあいだ,あるいは,発現しない骨髄マクロファージのあいだでの比較では,肥満あるいはアディポネクチンの有無はTNFの産生に影響しなかったことから,肥満マウスおよびアディポネクチンノックアウトマウスの骨髄においてはMHCクラスII分子を発現するM1様マクロファージの増加によりTNFの濃度が上昇すると考えられた.核内受容体型の転写因子であるNR4A1はM1様マクロファージの分化を抑制することが知られているが12),アディポネクチンはin vivoおよびin vitroにおいて単球におけるNR4A1の発現を誘導した.さらに,NR4A1のアゴニストの全身への投与により,骨髄マクロファージにおけるTNFの産生および造血前駆細胞におけるSOCS3の高発現が抑制された.
筆者らは,肥満モデルマウスにおける造血系の解析により,感染が重症化すること,それがアディポネクチンの欠乏による骨髄における炎症性サイトカインの亢進につづく顆粒球の産生の低下によること,アディポネクチンあるいはアディポネクチン受容体のアゴニストの投与によりこれらの異常が改善することを示した(図1).今回の研究は,肥満症に関連した感染症において,アディポネクチンが治療の標的となりうる可能性を示唆した.
略歴:2013年 東京大学大学院医学系研究科 修了,同年より東京大学医学部附属病院 特任助教(現 助教).
研究テーマ:骨髄系における正常な造血および腫瘍造血.
関心事:骨髄の環境が造血の機能にどのように影響をおよぼしているのか,それが白血病にどう関連しているのか,気になっています.
黒川 峰夫(Mineo Kurokawa)
東京大学大学院医学系研究科 教授.
研究室URL:http://www.u-tokyo-hemat.com/
© 2016 正本庸介・黒川峰夫 Licensed under CC 表示 2.1 日本
(東京大学大学院医学系研究科 血液・腫瘍病態学講座)
email:正本庸介,黒川峰夫
DOI: 10.7875/first.author.2016.069
Adiponectin enhances antibacterial activity of hematopoietic cells by suppressing bone marrow inflammation.
Yosuke Masamoto, Shunya Arai, Tomohiko Sato, Akihide Yoshimi, Naoto Kubota, Iseki Takamoto, Yoichiro Iwakura, Akihiko Yoshimura, Takashi Kadowaki, Mineo Kurokawa
Immunity, 44, 1422-1433 (2016)
要 約
肥満症によりさまざまな感染症の頻度および重症度の上がることが知られているが,その分子機序には不明な点が多い.今回,筆者らは,肥満モデルマウスの骨髄において,脂肪細胞の産生する“善玉”ホルモンであるアディポネクチンの濃度が低下し,それにともない炎症性サイトカインであるTNFの骨髄マクロファージからの分泌が慢性的に亢進することを示した.炎症性サイトカインに慢性的に曝露されることにより,骨髄の造血前駆細胞においてサイトカインシグナル抑制タンパク質であるSOCS3の発現が上昇し,細菌に感染した際のSTAT3シグナル伝達系の活性化が阻害される結果,感染からの防御に必須な顆粒球が骨髄において十分に産生されず細菌の駆逐が抑制された.肥満マウスにアディポネクチンあるいはアディポネクチン受容体のアゴニストであるAdipoRonを投与することにより,細菌に感染した際の顆粒球の産生は回復し重症化が抑制された.この結果から,アディポネクチンは肥満に関連した感染症の治療において標的になりうると考えられた.
はじめに
肥満症は心血管疾患,悪性腫瘍,感染症などさまざまな合併症と関連することが知られ,肥満症においてはさまざまな感染症の頻度および重症度が上がることが臨床的な研究により示唆されているが1),その分子機序には不明な点が多い.肥満症における中心的な病態は,脂肪細胞から分泌されるサイトカインであるアディポカインの,脂肪細胞の機能の異常にともなう産生の異常であると考えられている.肥満症においては脂肪細胞が肥大化しTNFやインターロイキン6などの炎症性サイトカインの産生が亢進する一方,アディポネクチンなど抗炎症作用をもつアディポカインの産生は逆説的に低下し,肥満症および合併症の病態を形成する.造血の主座である骨髄には脂肪細胞が豊富に存在し,肥満症における脂肪細胞の機能の異常は造血にもなんらかの影響をおよぼすと考えられるが,そのような検討はこれまでほとんどなされていない.今回,筆者らは,肥満症と感染症とのかかわりに注目し,肥満症における造血の異常が感染の重症化につながるという仮説をたてこれをマウスにおいて検証するとともに,肥満症における感染の重症化に対する治療的なアプローチについて検討した.
1.肥満マウスおよびアディポネクチンノックアウトマウスにおいて細菌に感染した際の顆粒球の産生が障害される
細菌の感染に対する適切な応答には,細菌を貪食する主体となる好中球が十分に産生されることが必要である.感染にともない内因性に産生されるサイトカインである顆粒球コロニー刺激因子は,骨髄における顆粒球前駆細胞の増殖および骨髄からの遊走を強く促進し,細菌の感染に対する応答にもっとも重要な役割をはたしており,顆粒球コロニー刺激因子を投与すると細菌に感染したときと類似した応答をひき起こすことが知られている2).汎用される肥満モデルマウスのひとつである高脂肪食を負荷したマウスに顆粒球コロニー刺激因子を投与すると,対照となるマウスにおいてみられた骨髄における顆粒球前駆細胞の十分な増殖がみられず,好中球の産生が低下した.
肥満モデルマウスの骨髄においてさまざまなサイトカインの発現を調べた研究において多くのサイトカインの発現量は変化しないことが報告されていたため3),多彩な生理的な機能をもつにもかかわらず,これまで,in vivoにおいて造血に対する作用が調べられていなかったアディポネクチンに注目した.肥満マウスにてアディポネクチンの血清における濃度は保たれていたが,骨髄における濃度は低下しており,骨髄においてアディポネクチンは血清とは異なる機構により制御されることが示唆された.
アディポネクチンノックアウトマウスは耐糖能の異常,動脈硬化への感受性の上昇などメタボリック症候群に類似した表現型を示すが,それらはいずれも軽微であることが知られている4).アディポネクチンノックアウトマウスの造血系には,細胞の数,分化,アポトーシス,細胞周期,造血幹細胞あるいは前駆細胞の活性を示す細胞の頻度など,さまざまな指標において明らかな異常はみられなかった.しかし,アディポネクチンノックアウトマウスに顆粒球コロニー刺激因子を投与したところ,肥満マウスと同様に,顆粒球前駆細胞の増殖の低下および好中球の産生の低下が認められた.
2.アディポネクチンが欠乏した状態において細菌の感染に対する応答が低下する
リステリア菌は細胞内寄生性のグラム陽性桿菌であり,おもに免疫不全者に対し重篤な全身感染症を起こす.リステリア菌の排除には顆粒球コロニー刺激因子によるシグナルの機能することが必要であることが示されており,リステリア菌の腹腔への感染はマウスにおける細菌感染モデルとして汎用される.この細菌感染モデルを用いて,肥満マウスおよびアディポネクチンノックアウトマウスの細菌の感染に対する応答を観察した.これらのマウスにおいては感染ののち骨髄および末梢血における好中球の増加,および,感染した部位への好中球の遊走が障害され,感染ののち7日目の肝臓および脾臓には有意に多数の細菌が残存しており感染は重症化していると考えられた.組換え型アディポネクチンの静脈内投与,あるいは,アディポネクチン受容体の小分子アゴニストであるAdipoRonの経口投与により,これらの感染に対する応答はいずれも回復し,細菌の数は低下した.
3.アディポネクチンの欠乏した状態の造血細胞において顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性が低下する
顆粒球コロニー刺激因子による顆粒球前駆細胞の増殖においては,STAT3シグナル伝達系の活性化が中心的な役割をはたすことが知られている.肥満マウスおよびアディポネクチンノックアウトマウスの造血細胞をin vitroにおいて顆粒球コロニー刺激因子により刺激したところ,対照となるマウスと比較して,STAT3のリン酸化が低下していた.そこで,これらのマウスにin vivoにおいて顆粒球コロニー刺激因子を投与したのち造血細胞を回収すると,STAT3のリン酸化が低下し,STAT3の下流の主要なエフェクターとして知られるC/EBPβの発現5) が低下していた.同様に,肥満マウスおよびアディポネクチンノックアウトマウスにおいて細菌の感染によりSTAT3シグナル伝達系の活性の低下がみられた.いずれのマウスにおいても,in vivoにおいてアディポネクチンを投与することによりSTAT3シグナル伝達系の活性化は回復した.
4.アディポネクチンの欠乏した状態では造血前駆細胞においてSOCS3が高発現し細菌に感染した際の顆粒球の産生が障害される
肥満マウスおよびアディポネクチンノックアウトマウスの造血細胞における顆粒球コロニー刺激因子に対する応答の低下の機序を明らかにするため,顆粒球コロニー刺激因子に応答した増殖の主体になると考えられる造血前駆細胞について詳細に解析した6).その結果,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化は,多能性前駆細胞,骨髄系共通前駆細胞,顆粒球マクロファージ前駆細胞といった前駆細胞の画分において顕著に低下していた.肥満モデルマウスの肝臓および筋肉においてSTAT3シグナル伝達系の主要な抑制タンパク質であるSOCS3の発現が上昇しインスリンのシグナルを抑制することが知られていたことから7),造血系においてSOCS3の発現を比較したところ,肥満マウスおよびアディポネクチンノックアウトマウスの造血前駆細胞においてSOCS3の発現が上昇し,in vivoにおけるアディポネクチンの投与によりこの発現は低下した.肥満マウスにおいて造血細胞の全体にてSOCS3の発現を低下させると,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化および細菌の感染に対する応答が回復した.一方で,肥満マウスにおいて成熟した顆粒球およびマクロファージにてSOCS3の発現を低下させても,これらの表現型に変化はみられなかった.以上のことから,アディポネクチンの欠乏した状態では造血前駆細胞におけるSOCS3の発現が上昇し,それによりSTAT3シグナル伝達系の活性化が抑制されると考えられた.
5.アディポネクチンの欠乏した状態では骨髄においてTNFの産生が亢進しSOCS3の高発現を誘導する
造血前駆細胞をin vitroにおいてアディポネクチンにより処理してもSOCS3の発現量に変化はみられなかった.そこで,生体においては,アディポネクチンにより発現が抑制されSOCS3の発現を誘導するタンパク質が作用するのではないかと考え,候補として,炎症性サイトカインであるTNFに注目した8-10).実際に,in vitroにおいてTNFは造血前駆細胞におけるSOCS3の発現を誘導し,また,肥満マウスおよびアディポネクチンノックアウトマウスの骨髄においてTNFの濃度が上昇しており,アディポネクチンの投与によりTNFの濃度は低下した.TNFノックアウトマウスにおいては肥満により造血前駆細胞におけるSOCS3の発現は上昇せず,顆粒球コロニー刺激因子に依存したSTAT3シグナル伝達系の活性化が回復したことから,TNFが重要なはたらきをすることが示唆された.
骨髄は血清と比較してTNFの濃度が高いことから,骨髄においては局所的にTNFが産生されると考えられた.骨髄のさまざまな細胞において比較したところ,多くの造血細胞の画分においてTNFの産生がみられたが,アディポネクチンによるTNFの産生の抑制はF4/80陽性のマクロファージでのみ観察された.そこで,薬理学的にマクロファージを消失させin vivoにおいて顆粒球コロニー刺激因子に対する応答を比較したところ,アディポネクチンノックアウトマウスにおいて細菌に感染した際の顆粒球の産生の低下はみられなくなり,アディポネクチンの作用はマクロファージを介することが示唆された.
6.アディポネクチンは骨髄においてTNFを産生するマクロファージの分化を抑制する
アディポネクチンが骨髄マクロファージにおいてTNFの産生を抑制する機構について検討した.さまざまな細胞種を用いた実験においてアディポネクチンはリポ多糖により刺激したときのTNFの発現を転写レベルにおいて直接的に抑制することが知られているが11),生体のように,リポ多糖のようなTNFを産生させる強力な刺激が存在せず,長時間にわたりアディポネクチンに曝露される状態において,アディポネクチンがTNFの産生にどのように影響するかは不明であった.そこで,アディポネクチンノックアウトマウスの骨髄からマクロファージを単離し,リポ多糖の添加あるいは非添加の条件においてアディポネクチンを処理したところ,アディポネクチンはリポ多糖により刺激したときのTNFの発現を転写レベルで強力に抑制したものの,リポ多糖の刺激のないときのTNFの発現に対する抑制の効果は比較的小さかった.そのため,アディポネクチンが骨髄マクロファージに直接的に作用してTNFの産生を抑制する以外にも,なんらかの機序の存在が示唆された.
おもにin vitroにおける実験において,マクロファージは周囲のサイトカインに応答して炎症性のM1マクロファージあるいは抗炎症性のM2マクロファージに分化することが知られている.そこで,肥満マウスおよびアディポネクチンノックアウトマウスから骨髄マクロファージを単離して遺伝子の発現を比較したところ,TNF遺伝子を含むM1マクロファージと関連する複数の遺伝子の発現の上昇がみられ,また,M1マクロファージの形質と関連することの知られているMHCクラスII分子を発現するマクロファージが増加した.肥満マウス,アディポネクチンノックアウトマウス,対照となるマウスのいずれにおいても,MHCクラスII分子を発現する骨髄マクロファージはTNFを多く産生しており,MHCクラスII分子を発現する骨髄マクロファージのあいだ,あるいは,発現しない骨髄マクロファージのあいだでの比較では,肥満あるいはアディポネクチンの有無はTNFの産生に影響しなかったことから,肥満マウスおよびアディポネクチンノックアウトマウスの骨髄においてはMHCクラスII分子を発現するM1様マクロファージの増加によりTNFの濃度が上昇すると考えられた.核内受容体型の転写因子であるNR4A1はM1様マクロファージの分化を抑制することが知られているが12),アディポネクチンはin vivoおよびin vitroにおいて単球におけるNR4A1の発現を誘導した.さらに,NR4A1のアゴニストの全身への投与により,骨髄マクロファージにおけるTNFの産生および造血前駆細胞におけるSOCS3の高発現が抑制された.
おわりに
筆者らは,肥満モデルマウスにおける造血系の解析により,感染が重症化すること,それがアディポネクチンの欠乏による骨髄における炎症性サイトカインの亢進につづく顆粒球の産生の低下によること,アディポネクチンあるいはアディポネクチン受容体のアゴニストの投与によりこれらの異常が改善することを示した(図1).今回の研究は,肥満症に関連した感染症において,アディポネクチンが治療の標的となりうる可能性を示唆した.
文 献
- Falagas, M. E. & Kompoti, M.: Obesity and infection. Lancet Infect. Dis., 6, 438-446 (2006)[PubMed]
- Boettcher, S., Ziegler, P., Schmid, M. A. et al.: Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J. Immunol., 188, 5824-5828 (2012)[PubMed]
- Trottier, M. D., Naaz, A., Li, Y. et al.: Enhancement of hematopoiesis and lymphopoiesis in diet-induced obese mice. Proc. Natl. Acad. Sci. USA, 109, 7622-7629 (2012)[PubMed]
- Kubota, N., Terauchi, Y., Yamauchi, T. et al.: Disruption of adiponectin causes insulin resistance and neointimal formation. J. Biol. Chem., 277, 25863-25866 (2002)[PubMed]
- Hirai, H., Zhang, P., Dayaram, T. et al.: C/EBPβ is required for 'emergency' granulopoiesis. Nat. Immunol., 7, 732-739 (2006)[PubMed]
- Zhang, H., Nguyen-Jackson, H., Panopoulos, A. D. et al.: STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood, 116, 2462-2471 (2010)[PubMed]
- Ueki, K., Kondo, T. & Kahn, C. R.: Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol., 24, 5434-5446 (2004)[PubMed]
- Maeda, N., Shimomura, I., Kishida, K. et al.: Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med., 8, 731-737 (2002)[PubMed]
- Weisberg, S. P., McCann, D., Desai, M. et al.: Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest., 112, 1796-1808 (2003)[PubMed]
- Ehlting, C., Lai, W. S., Schaper, F. et al.: Regulation of suppressor of cytokine signaling 3 (SOCS3) mRNA stability by TNF-α involves activation of the MKK6/p38MAPK/MK2 cascade. J. Immunol., 178, 2813-2826 (2007)[PubMed]
- Park, P. H., Huang, H., McMullen, M. R. et al.: Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J. Biol. Chem., 283, 26850-26858 (2008)[PubMed]
- Hanna, R. N., Shaked, I., Hubbeling, H. G. et al.: NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res., 110, 416-427 (2012)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2013年 東京大学大学院医学系研究科 修了,同年より東京大学医学部附属病院 特任助教(現 助教).
研究テーマ:骨髄系における正常な造血および腫瘍造血.
関心事:骨髄の環境が造血の機能にどのように影響をおよぼしているのか,それが白血病にどう関連しているのか,気になっています.
黒川 峰夫(Mineo Kurokawa)
東京大学大学院医学系研究科 教授.
研究室URL:http://www.u-tokyo-hemat.com/
© 2016 正本庸介・黒川峰夫 Licensed under CC 表示 2.1 日本
