治療に抵抗性の変異を克服する新世代のmTOR阻害薬の創出
岡庭正格・Kevan M. Shokat
(米国California大学San Francisco校Department of Cellular and Molecular Pharmacology)
email:岡庭正格
DOI: 10.7875/first.author.2016.062
Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor.
Vanessa S. Rodrik-Outmezguine, Masanori Okaniwa, Zhan Yao, Chris J. Novotny, Claire McWhirter, Arpitha Banaji, Helen Won, Wai Wong, Mike Berger, Elisa de Stanchina, Derek G. Barratt, Sabina Cosulich, Teresa Klinowska, Neal Rosen, Kevan M. Shokat
Nature, 534, 272-276 (2016)
がん治療における精密治療は,治療の環境に適応した薬剤に抵抗性のがん細胞を選択的に生き残らせてしまい,薬物治療の初期には効果が得られたとしても,のちに治療に抵抗性のがんが再発するリスクがあり,これを克服するため次世代の治療法の開発がもとめられている.mTORシグナル伝達経路はがんにおいて高頻度に活性化されるシグナル伝達経路のひとつであり,このシグナル伝達経路を標的とした創薬がさかんに行われてきた.筆者らは,mTORキナーゼ阻害薬による治療歴のないがん患者においてmTORに活性化型の変異が存在し,この変異がmTORキナーゼ阻害薬への治療の抵抗性の原因になることを明らかにした.さらに,第1世代のmTOR阻害薬であるラパマイシン誘導体と第2世代のmTOR阻害薬であるmTORキナーゼ阻害薬をリンカーで結合させ,mTORの2つの異なる薬剤結合部位に同時に結合できるよう最適化したmTOR阻害薬としてRapaLink-1を開発した.このドラッグデザインが既存のmTOR阻害薬への耐性を克服する有効な手段となった.
現状,がんにおける精密治療(precision medicine)は究極の目標であるがんの克服にはいたっていない.今後,ゲノム情報の蓄積および治療薬の分類により,個別の患者にあった治療薬を選択できるようにする必要がある.それにくわえ,薬物治療の初期には効果が得られたとしても,がんが完全に消失しないかぎり,のちに治療に抵抗性のがんが再発するリスクがある.必ず生じるであろう薬剤耐性をいかにして克服するかが,がん治療において解決できていない大きな課題である.
ラパマイシンはイースター島の土壌から単離された放線菌の産生するマクロライド系天然物として発見され,免疫抑制の活性をもつことから臓器移植における拒絶反応を抑制する薬剤として使用されるようになった.さらに,ラパマイシンの標的となるタンパク質がmTORであること,また,抗がん活性を示すことが明らかにされ,第1世代のmTOR阻害薬としてラパマイシン誘導体の研究開発がさかんに行われてきた.ラパマイシンは細胞に存在するタンパク質FKBP12と結合したのち,mTORのFRBドメインとも結合し,ラパマイシン,FKBP12,mTORからなる安定な三者複合体を形成することがX線結晶構造解析により明らかにされた1).ラパマイシンの作用機序は,mTORの活性中心の近傍にリクルートされたFKBP12の立体障害によりリン酸化されるべき基質が近づけなくなることだと考えられている2).そのため,ラパマイシン誘導体によるmTORの阻害活性の強度は基質の種類により異なり,mTOR複合体1のエフェクタータンパク質であるS6KやS6などを強く抑制する.
第2世代のmTOR阻害薬であるmTORキナーゼ阻害薬は,リン酸基の転移反応に用いられるATPの構造を模倣した低分子化合物であり,細胞においてATPと拮抗しながらmTORの活性中心に結合しそのキナーゼ活性を抑制する.この分子機構により,mTORキナーゼ阻害薬はmTOR複合体1およびmTOR複合体2のすべての基質に対するリン酸化反応を低下させる.そのため,mTORキナーゼ阻害薬の抗がん作用はラパマイシン誘導体よりも強力であるとみこまれる.mTORはがん細胞だけではなく正常な細胞にも存在することから,安全性については慎重に検討される必要がある.現在,複数のmTORキナーゼ阻害薬が臨床試験により評価中である.
ヒトの乳がん細胞であるMCF-7細胞を3カ月間にわたり高濃度の第1世代のラパマイシン誘導体あるいは第2世代のmTORキナーゼ阻害薬に曝露させることにより,それぞれの薬剤耐性株が樹立された.遺伝子解析の結果,mTORキナーゼ阻害薬に耐性のクローンはmTORのキナーゼドメインに変異をもち,ラパマイシン誘導体に耐性のクローンはmTORのFRBドメインに変異をもつことが明らかにされた.これらの獲得変異の臨床における妥当性を示唆するエビデンスとして,第1世代のmTOR阻害薬であるEverolimusによる処置のもとで再発したがん患者において,mTORのFRBドメインに同じ変異が報告されている3).
これらの細胞を用いて,mTORシグナル伝達経路を構成するタンパク質のリン酸化の状態を解析した.S6KおよびS6はラパマイシン誘導体によりリン酸化が抑制されるが,ラパマイシン誘導体に耐性の細胞においてはラパマイシン誘導体を処理してもリン酸化された.一方,ラパマイシン誘導体に耐性の細胞にmTORキナーゼ阻害薬を処理した場合にはS6KおよびS6のリン酸化は抑制され,さらに,mTOR複合体1の別のエフェクタータンパク質である4EBP1のリン酸化も抑制された.mTORキナーゼ阻害薬に耐性の細胞にmTORキナーゼ阻害薬を処理したところ,S6KおよびS6のリン酸化の抑制は明らかに減弱した.mTORキナーゼ阻害薬に耐性の細胞にラパマイシン誘導体を処理した場合には,S6KおよびS6のリン酸化のみが抑制され,4EBP1のリン酸化は抑制されなかった.細胞増殖試験においては,これらのシグナル伝達経路の応答性を反映して,ラパマイシン誘導体に耐性の細胞はラパマイシン誘導体に対し,mTORキナーゼ阻害薬に耐性の細胞はmTORキナーゼ阻害薬に対し,顕著に低い感受性を示した.mTORの変異が薬剤抵抗性の直接の原因となることを証明するため,それぞれの変異型mTORをMDA-MB-468細胞に発現させ,同様の薬剤抵抗性の生じることが確認された.
ラパマイシン誘導体に耐性の変異は,FRBドメインに生じた1アミノ酸の変異がmTORとFKBP12-ラパマイシン複合体との相互作用をさまたげるという,出芽酵母において知られている耐性の機構と同様と考えられた4).一方,mTORキナーゼ阻害薬に耐性の変異はmTORキナーゼ阻害薬の結合ポケットから15Å以上も離れた部位に存在しており,薬剤の結合をアロステリックに制御するのか,あるいは,薬剤の結合とは関係のない機構が存在するのか検証する必要があった.野生型のmTORおよびmTORキナーゼ阻害薬に耐性のmTORはmTORキナーゼ阻害薬に対し同じ程度の親和性を示したが,mTORキナーゼ阻害薬に耐性のmTORは野生型のmTORと比べ3倍も高いキナーゼ活性を示した.すなわち,このmTORの変異は活性化型であった.
このmTORの活性化型の変異はmTORキナーゼ阻害薬による治療歴のないがん患者にも存在することが報告されていたため5,6),ほかの4つのキナーゼドメインの変異についても薬剤感受性の低下およびキナーゼ活性の活性化について検証した.MDA-MB-468細胞にこれらの変異をもつmTORを発現させmTORキナーゼ阻害薬を処理したところ,mTOR複合体1あるいはmTOR複合体2の基質のリン酸化を抑制するには3~30倍の濃度が必要であった.mTORの活性化型の変異はmTORキナーゼ阻害薬による治療歴のない場合にもmTORキナーゼ阻害薬による治療に対し抵抗性をもつというエビデンスは,次世代のmTOR阻害薬の必要性を強く問いかけている.
2013年,mTORキナーゼ阻害薬とmTORとの複合体のX線結晶構造が報告された7).この発見により,ラパマイシン誘導体の結合部位とmTORキナーゼ阻害薬の結合部位の位置の関係がより明確になり,2つの薬剤結合部位と同時に結合することのできる新世代のmTOR阻害薬RapaLinkをデザインすることが可能になった(図1a).薬剤耐性を克服する手段のひとつとして,2つの薬剤を適切なリンカーで結合し,ひとつの標的タンパク質のもつそれぞれの薬剤結合部位に2価の相互作用で強力に結合するという,あたかも抗体のようなアビディティ効果にもとづくアプローチを考案した.すなわち,FRBドメインの変異にもとづくラパマイシン誘導体に対する耐性にはmTORキナーゼ阻害薬との結合部位の結合力により,キナーゼドメインの変異にもとづくmTORキナーゼ阻害薬に対する耐性には2価の強力な結合力によりmTORの機能を阻害できるのではないだろうか.
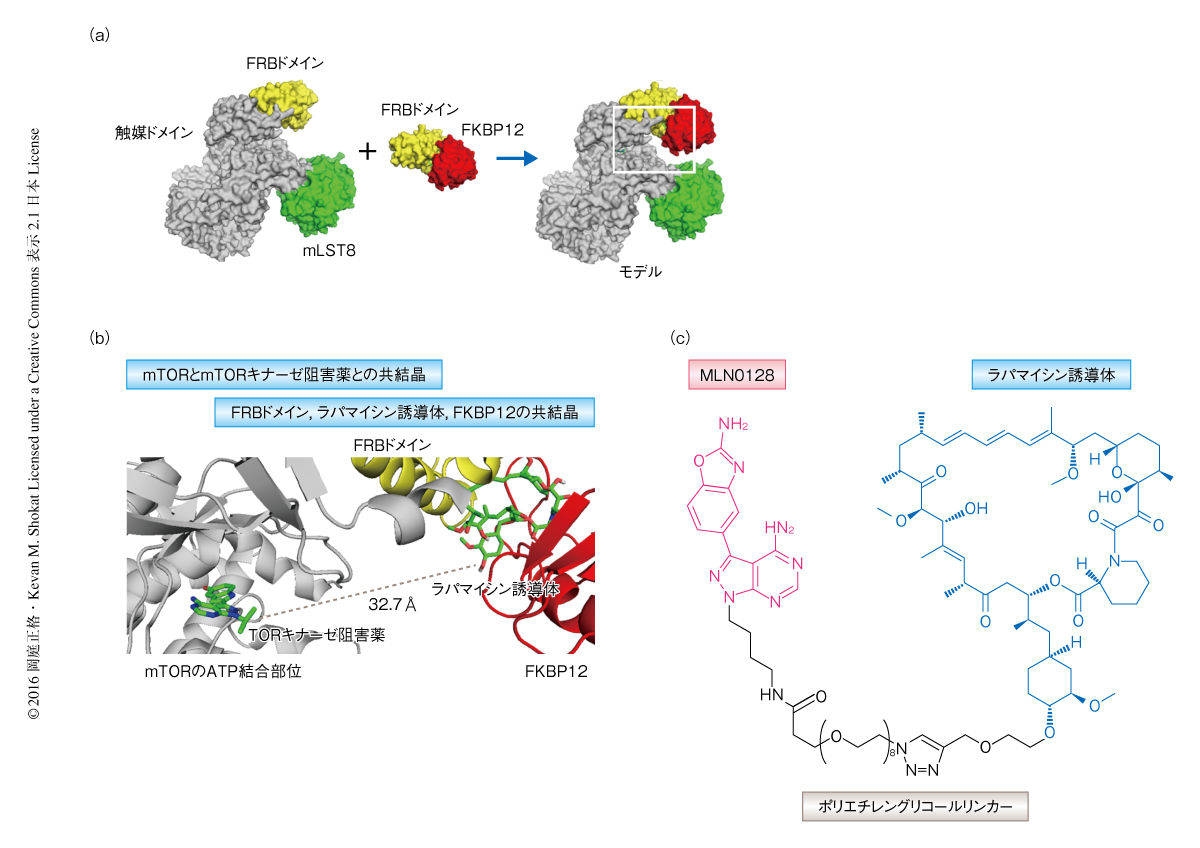
分子モデルを用いた解析により,ラパマイシン誘導体のC40位の水酸基はmTORのキナーゼドメインにむかい配向し,一方のmTORキナーゼ阻害薬のもつ縮合ピラゾール環のN-1位はラパマイシン誘導体にむかい配向していることが明らかにされた(図1b).そこで,mTORキナーゼ阻害薬としてmTORに高選択的かつ臨床試験が進行中の縮合ピラゾール誘導体MLN0128を選択した8).適切なリンカー長を予測するためエネルギー計算を行ったところ,ラパマイシン誘導体およびMLN0128の2つの要素を大きなひずみなく結合し,かつ,おのおのの結合部位と同時に相互作用させるために必要なリンカー長は27重原子長より長い必要のあることが判明した.そこで,さまざまな重原子長をもつポリエチレングリコールリンカーによりラパマイシン誘導体とMLN0128を結合した誘導体として,39重原子長のRapaLink-1および36重原子長のRapaLink-2,さらに,11重原子長の理論的に2価の結合のできない陰性対照としてRapaLink-3を合成した(図1c).
RapaLink-1,RapaLink-2,RapaLink-3をMCF-7細胞に作用させ,mTORシグナル伝達経路を構成するタンパク質をウェスタンブロット法により解析した結果,RapaLink-1およびRapaLink-2はmTOR複合体1あるいはmTOR複合体2の下流タンパク質のリン酸化を1~3 nMの濃度で阻害した.しかしながら,RapaLink-3はS6に対するリン酸化の阻害活性は保持したものの,4EBP1およびAKTに対するリン酸化の阻害活性は減弱した.これらの結果はエネルギー計算の結果と一致しており,かつ,S6に対する活性が4EBP1に対する活性よりも優勢であったことから,MLN0128の結合力よりもラパマイシン誘導体の結合力が優勢であることが示された.RapaLink-1はMCF-7細胞に対し強力な増殖の阻害活性を示した.
治療薬として2価で結合する中程度の分子量をもつ化合物のデザインはこれまでも報告されていたが,ハイブリッド化合物の薬理学的な特性により成否を分けてきた.とりわけ,ラパマイシン誘導体などFKBP12と結合する化合物はハイブリッド化合物の薬理学的な特性を向上させるため利用されている.たとえば,FK506をベースとするハイブリッド化合物は,FK506とFKBP12との高い親和性と,とくに赤血球におけるFKBP12の高い濃度を利用して,薬物のリザーバーをつくりだし血中における半減期の延長が得られる9).これらの知見と一致して,RapaLink-1は薬物のウォッシュアウト試験においてMLN0128と比較して有意に長いmTORシグナル伝達経路の阻害作用の持続が確認された.ラパマイシン誘導体も同様に阻害作用が持続することから,これはラパマイシン誘導体による効果と考えられた.
ヒトの乳がん細胞であるMCF-7細胞を移植したマウスモデルにおいて,RapaLink-1は単回の投与ののち4日間にわたり,腫瘍においてmTOR複合体1あるいはmTOR複合体2の下流タンパク質を抑制した.さらに,RapaLink-1はラパマイシン誘導体やmTORキナーゼ阻害薬と同等の抗腫瘍効果を示した.このように強力なmTOR阻害活性をもつRapaLink-1の安全な投与量は臨床試験により詳細に検討する必要がある.
RapaLink-1の薬剤耐性変異に対する作用を評価するため,MDA-MB-468細胞にラパマイシン誘導体に耐性の変異をもつmTORあるいはmTORキナーゼ阻害薬に耐性の変異をもつmTORを発現させ,ラパマイシン誘導体,MLN0128,あるいは,その併用の効果と比較した.その結果,RapaLink-1のみが3~10 nMという低濃度でmTORシグナル伝達経路を阻害した(図2).さらに,ヒトの腫瘍を移植したマウスモデルにおいて,ラパマイシン誘導体に耐性の変異をもつmTORを発現させたMCF-7細胞は,ラパマイシン誘導体への感受性が低下したものの,mTORキナーゼ阻害薬およびRapaLink-1に対し高い感受性を示した.同様に,mTORキナーゼ阻害薬に耐性の変異をもつmTORを発現させたMCF-7細胞は,mTORキナーゼ阻害薬への感受性が低下したものの,ラパマイシン誘導体およびRapaLink-1には同様に高い感受性を保持した.
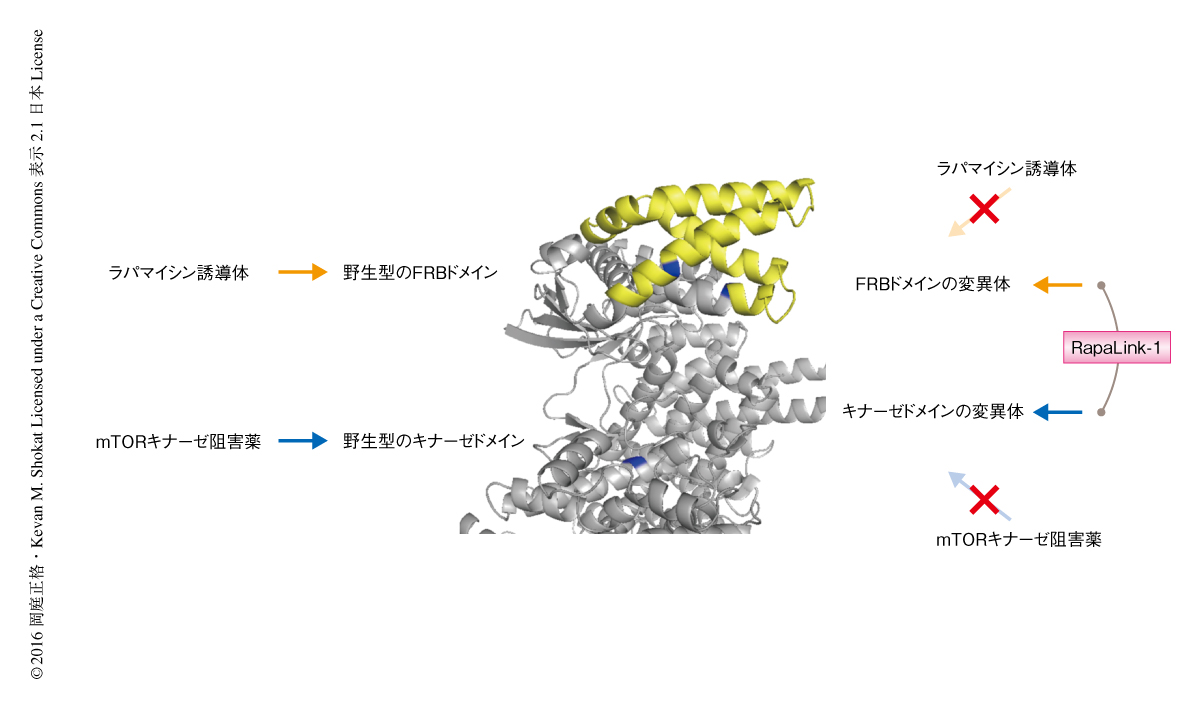
mTORのキナーゼドメインに活性化型の変異をもつ患者はラパマイシン誘導体に応答性を示すが,治療の開始ののち2つ目の耐性変異が生じると多剤耐性を獲得してしまう.このような獲得耐性を想定して,FRBドメインおよびキナーゼドメインに変異をもつmTORをMDA-MB-468細胞に発現させRapaLink-1の効果を検討した.この二重変異mTORは想定どおりラパマイシン誘導体およびmTORキナーゼ阻害薬,さらに,ラパマイシン誘導体とmTORキナーゼ阻害薬の併用に耐性を示したのに対し,RapaLink-1に対しては感受性を示した.
筆者らは,mTORのキナーゼドメインとFRBドメインの両方を標的として同時に結合することができる新世代のmTOR阻害薬RapaLinkを創出した.筆者らが提唱した1分子のmTORに対しRapaLink-1が2座配位するという説は,最近,クライオ電子顕微鏡により解明されたmTOR複合体1の二量体の構造において,2分子のmTORのあいだの距離がRapaLink-1の分子長よりも有意に離れていることからも支持された2).RapaLink-1の片側の要素がmTORと結合すると,適切なリンカー長で結合したもう片方の要素が結合部位の近傍にとどまり,強い結合親和力が得られる.RapaLink-1に対する耐性株を樹立するためには,ラパマイシン誘導体あるいはmTORキナーゼ阻害薬の3カ月と比較して9カ月という長い期間を必要としたことから,RapaLink-1はがん細胞が薬剤耐性を獲得するまでの期間を延長できるかもしれない.このように,新世代のmTOR阻害薬であるRapaLink-1の創出をつうじ,がんの耐性の克服への新しいアプローチが提案された.
略歴:2013年 京都薬科大学大学院にて博士号取得,同年 米国California大学San Francisco校 博士研究員を経て,2014年 武田薬品工業,2016年より同 主席研究員.
研究テーマ:ケミカルバイオロジーとメディシナルケミストリーをベースにした創薬.
抱負:すぐれた医薬品の創出に挑戦し,創薬のプロフェッショナルとして成長して,医療の未来に貢献したい.
Kevan M. Shokat
米国California大学San Francisco校 教授.
研究室URL:http://shokatlab.ucsf.edu/
© 2016 岡庭正格・Kevan M. Shokat Licensed under CC 表示 2.1 日本
(米国California大学San Francisco校Department of Cellular and Molecular Pharmacology)
email:岡庭正格
DOI: 10.7875/first.author.2016.062
Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor.
Vanessa S. Rodrik-Outmezguine, Masanori Okaniwa, Zhan Yao, Chris J. Novotny, Claire McWhirter, Arpitha Banaji, Helen Won, Wai Wong, Mike Berger, Elisa de Stanchina, Derek G. Barratt, Sabina Cosulich, Teresa Klinowska, Neal Rosen, Kevan M. Shokat
Nature, 534, 272-276 (2016)
この論文に出現する遺伝子・タンパク質のUniprot ID
mTOR(P42345), mTORキナーゼ, FKBP12(P62942), mTOR複合体1, S6K(P23443), S6(P62753), mTOR複合体2, 4EBP1(Q13541), AKT(P31749)
要 約
がん治療における精密治療は,治療の環境に適応した薬剤に抵抗性のがん細胞を選択的に生き残らせてしまい,薬物治療の初期には効果が得られたとしても,のちに治療に抵抗性のがんが再発するリスクがあり,これを克服するため次世代の治療法の開発がもとめられている.mTORシグナル伝達経路はがんにおいて高頻度に活性化されるシグナル伝達経路のひとつであり,このシグナル伝達経路を標的とした創薬がさかんに行われてきた.筆者らは,mTORキナーゼ阻害薬による治療歴のないがん患者においてmTORに活性化型の変異が存在し,この変異がmTORキナーゼ阻害薬への治療の抵抗性の原因になることを明らかにした.さらに,第1世代のmTOR阻害薬であるラパマイシン誘導体と第2世代のmTOR阻害薬であるmTORキナーゼ阻害薬をリンカーで結合させ,mTORの2つの異なる薬剤結合部位に同時に結合できるよう最適化したmTOR阻害薬としてRapaLink-1を開発した.このドラッグデザインが既存のmTOR阻害薬への耐性を克服する有効な手段となった.
はじめに
現状,がんにおける精密治療(precision medicine)は究極の目標であるがんの克服にはいたっていない.今後,ゲノム情報の蓄積および治療薬の分類により,個別の患者にあった治療薬を選択できるようにする必要がある.それにくわえ,薬物治療の初期には効果が得られたとしても,がんが完全に消失しないかぎり,のちに治療に抵抗性のがんが再発するリスクがある.必ず生じるであろう薬剤耐性をいかにして克服するかが,がん治療において解決できていない大きな課題である.
1.mTOR阻害薬の分類
ラパマイシンはイースター島の土壌から単離された放線菌の産生するマクロライド系天然物として発見され,免疫抑制の活性をもつことから臓器移植における拒絶反応を抑制する薬剤として使用されるようになった.さらに,ラパマイシンの標的となるタンパク質がmTORであること,また,抗がん活性を示すことが明らかにされ,第1世代のmTOR阻害薬としてラパマイシン誘導体の研究開発がさかんに行われてきた.ラパマイシンは細胞に存在するタンパク質FKBP12と結合したのち,mTORのFRBドメインとも結合し,ラパマイシン,FKBP12,mTORからなる安定な三者複合体を形成することがX線結晶構造解析により明らかにされた1).ラパマイシンの作用機序は,mTORの活性中心の近傍にリクルートされたFKBP12の立体障害によりリン酸化されるべき基質が近づけなくなることだと考えられている2).そのため,ラパマイシン誘導体によるmTORの阻害活性の強度は基質の種類により異なり,mTOR複合体1のエフェクタータンパク質であるS6KやS6などを強く抑制する.
第2世代のmTOR阻害薬であるmTORキナーゼ阻害薬は,リン酸基の転移反応に用いられるATPの構造を模倣した低分子化合物であり,細胞においてATPと拮抗しながらmTORの活性中心に結合しそのキナーゼ活性を抑制する.この分子機構により,mTORキナーゼ阻害薬はmTOR複合体1およびmTOR複合体2のすべての基質に対するリン酸化反応を低下させる.そのため,mTORキナーゼ阻害薬の抗がん作用はラパマイシン誘導体よりも強力であるとみこまれる.mTORはがん細胞だけではなく正常な細胞にも存在することから,安全性については慎重に検討される必要がある.現在,複数のmTORキナーゼ阻害薬が臨床試験により評価中である.
2.mTOR阻害薬に対する耐性株の樹立とmTORの変異および耐性化の機構
ヒトの乳がん細胞であるMCF-7細胞を3カ月間にわたり高濃度の第1世代のラパマイシン誘導体あるいは第2世代のmTORキナーゼ阻害薬に曝露させることにより,それぞれの薬剤耐性株が樹立された.遺伝子解析の結果,mTORキナーゼ阻害薬に耐性のクローンはmTORのキナーゼドメインに変異をもち,ラパマイシン誘導体に耐性のクローンはmTORのFRBドメインに変異をもつことが明らかにされた.これらの獲得変異の臨床における妥当性を示唆するエビデンスとして,第1世代のmTOR阻害薬であるEverolimusによる処置のもとで再発したがん患者において,mTORのFRBドメインに同じ変異が報告されている3).
これらの細胞を用いて,mTORシグナル伝達経路を構成するタンパク質のリン酸化の状態を解析した.S6KおよびS6はラパマイシン誘導体によりリン酸化が抑制されるが,ラパマイシン誘導体に耐性の細胞においてはラパマイシン誘導体を処理してもリン酸化された.一方,ラパマイシン誘導体に耐性の細胞にmTORキナーゼ阻害薬を処理した場合にはS6KおよびS6のリン酸化は抑制され,さらに,mTOR複合体1の別のエフェクタータンパク質である4EBP1のリン酸化も抑制された.mTORキナーゼ阻害薬に耐性の細胞にmTORキナーゼ阻害薬を処理したところ,S6KおよびS6のリン酸化の抑制は明らかに減弱した.mTORキナーゼ阻害薬に耐性の細胞にラパマイシン誘導体を処理した場合には,S6KおよびS6のリン酸化のみが抑制され,4EBP1のリン酸化は抑制されなかった.細胞増殖試験においては,これらのシグナル伝達経路の応答性を反映して,ラパマイシン誘導体に耐性の細胞はラパマイシン誘導体に対し,mTORキナーゼ阻害薬に耐性の細胞はmTORキナーゼ阻害薬に対し,顕著に低い感受性を示した.mTORの変異が薬剤抵抗性の直接の原因となることを証明するため,それぞれの変異型mTORをMDA-MB-468細胞に発現させ,同様の薬剤抵抗性の生じることが確認された.
ラパマイシン誘導体に耐性の変異は,FRBドメインに生じた1アミノ酸の変異がmTORとFKBP12-ラパマイシン複合体との相互作用をさまたげるという,出芽酵母において知られている耐性の機構と同様と考えられた4).一方,mTORキナーゼ阻害薬に耐性の変異はmTORキナーゼ阻害薬の結合ポケットから15Å以上も離れた部位に存在しており,薬剤の結合をアロステリックに制御するのか,あるいは,薬剤の結合とは関係のない機構が存在するのか検証する必要があった.野生型のmTORおよびmTORキナーゼ阻害薬に耐性のmTORはmTORキナーゼ阻害薬に対し同じ程度の親和性を示したが,mTORキナーゼ阻害薬に耐性のmTORは野生型のmTORと比べ3倍も高いキナーゼ活性を示した.すなわち,このmTORの変異は活性化型であった.
このmTORの活性化型の変異はmTORキナーゼ阻害薬による治療歴のないがん患者にも存在することが報告されていたため5,6),ほかの4つのキナーゼドメインの変異についても薬剤感受性の低下およびキナーゼ活性の活性化について検証した.MDA-MB-468細胞にこれらの変異をもつmTORを発現させmTORキナーゼ阻害薬を処理したところ,mTOR複合体1あるいはmTOR複合体2の基質のリン酸化を抑制するには3~30倍の濃度が必要であった.mTORの活性化型の変異はmTORキナーゼ阻害薬による治療歴のない場合にもmTORキナーゼ阻害薬による治療に対し抵抗性をもつというエビデンスは,次世代のmTOR阻害薬の必要性を強く問いかけている.
3.mTORの2つの薬剤標的部位と同時に結合する新規の薬剤RapaLinkの創出
2013年,mTORキナーゼ阻害薬とmTORとの複合体のX線結晶構造が報告された7).この発見により,ラパマイシン誘導体の結合部位とmTORキナーゼ阻害薬の結合部位の位置の関係がより明確になり,2つの薬剤結合部位と同時に結合することのできる新世代のmTOR阻害薬RapaLinkをデザインすることが可能になった(図1a).薬剤耐性を克服する手段のひとつとして,2つの薬剤を適切なリンカーで結合し,ひとつの標的タンパク質のもつそれぞれの薬剤結合部位に2価の相互作用で強力に結合するという,あたかも抗体のようなアビディティ効果にもとづくアプローチを考案した.すなわち,FRBドメインの変異にもとづくラパマイシン誘導体に対する耐性にはmTORキナーゼ阻害薬との結合部位の結合力により,キナーゼドメインの変異にもとづくmTORキナーゼ阻害薬に対する耐性には2価の強力な結合力によりmTORの機能を阻害できるのではないだろうか.
分子モデルを用いた解析により,ラパマイシン誘導体のC40位の水酸基はmTORのキナーゼドメインにむかい配向し,一方のmTORキナーゼ阻害薬のもつ縮合ピラゾール環のN-1位はラパマイシン誘導体にむかい配向していることが明らかにされた(図1b).そこで,mTORキナーゼ阻害薬としてmTORに高選択的かつ臨床試験が進行中の縮合ピラゾール誘導体MLN0128を選択した8).適切なリンカー長を予測するためエネルギー計算を行ったところ,ラパマイシン誘導体およびMLN0128の2つの要素を大きなひずみなく結合し,かつ,おのおのの結合部位と同時に相互作用させるために必要なリンカー長は27重原子長より長い必要のあることが判明した.そこで,さまざまな重原子長をもつポリエチレングリコールリンカーによりラパマイシン誘導体とMLN0128を結合した誘導体として,39重原子長のRapaLink-1および36重原子長のRapaLink-2,さらに,11重原子長の理論的に2価の結合のできない陰性対照としてRapaLink-3を合成した(図1c).
RapaLink-1,RapaLink-2,RapaLink-3をMCF-7細胞に作用させ,mTORシグナル伝達経路を構成するタンパク質をウェスタンブロット法により解析した結果,RapaLink-1およびRapaLink-2はmTOR複合体1あるいはmTOR複合体2の下流タンパク質のリン酸化を1~3 nMの濃度で阻害した.しかしながら,RapaLink-3はS6に対するリン酸化の阻害活性は保持したものの,4EBP1およびAKTに対するリン酸化の阻害活性は減弱した.これらの結果はエネルギー計算の結果と一致しており,かつ,S6に対する活性が4EBP1に対する活性よりも優勢であったことから,MLN0128の結合力よりもラパマイシン誘導体の結合力が優勢であることが示された.RapaLink-1はMCF-7細胞に対し強力な増殖の阻害活性を示した.
4.RapaLink-1の薬効の評価
治療薬として2価で結合する中程度の分子量をもつ化合物のデザインはこれまでも報告されていたが,ハイブリッド化合物の薬理学的な特性により成否を分けてきた.とりわけ,ラパマイシン誘導体などFKBP12と結合する化合物はハイブリッド化合物の薬理学的な特性を向上させるため利用されている.たとえば,FK506をベースとするハイブリッド化合物は,FK506とFKBP12との高い親和性と,とくに赤血球におけるFKBP12の高い濃度を利用して,薬物のリザーバーをつくりだし血中における半減期の延長が得られる9).これらの知見と一致して,RapaLink-1は薬物のウォッシュアウト試験においてMLN0128と比較して有意に長いmTORシグナル伝達経路の阻害作用の持続が確認された.ラパマイシン誘導体も同様に阻害作用が持続することから,これはラパマイシン誘導体による効果と考えられた.
ヒトの乳がん細胞であるMCF-7細胞を移植したマウスモデルにおいて,RapaLink-1は単回の投与ののち4日間にわたり,腫瘍においてmTOR複合体1あるいはmTOR複合体2の下流タンパク質を抑制した.さらに,RapaLink-1はラパマイシン誘導体やmTORキナーゼ阻害薬と同等の抗腫瘍効果を示した.このように強力なmTOR阻害活性をもつRapaLink-1の安全な投与量は臨床試験により詳細に検討する必要がある.
RapaLink-1の薬剤耐性変異に対する作用を評価するため,MDA-MB-468細胞にラパマイシン誘導体に耐性の変異をもつmTORあるいはmTORキナーゼ阻害薬に耐性の変異をもつmTORを発現させ,ラパマイシン誘導体,MLN0128,あるいは,その併用の効果と比較した.その結果,RapaLink-1のみが3~10 nMという低濃度でmTORシグナル伝達経路を阻害した(図2).さらに,ヒトの腫瘍を移植したマウスモデルにおいて,ラパマイシン誘導体に耐性の変異をもつmTORを発現させたMCF-7細胞は,ラパマイシン誘導体への感受性が低下したものの,mTORキナーゼ阻害薬およびRapaLink-1に対し高い感受性を示した.同様に,mTORキナーゼ阻害薬に耐性の変異をもつmTORを発現させたMCF-7細胞は,mTORキナーゼ阻害薬への感受性が低下したものの,ラパマイシン誘導体およびRapaLink-1には同様に高い感受性を保持した.
mTORのキナーゼドメインに活性化型の変異をもつ患者はラパマイシン誘導体に応答性を示すが,治療の開始ののち2つ目の耐性変異が生じると多剤耐性を獲得してしまう.このような獲得耐性を想定して,FRBドメインおよびキナーゼドメインに変異をもつmTORをMDA-MB-468細胞に発現させRapaLink-1の効果を検討した.この二重変異mTORは想定どおりラパマイシン誘導体およびmTORキナーゼ阻害薬,さらに,ラパマイシン誘導体とmTORキナーゼ阻害薬の併用に耐性を示したのに対し,RapaLink-1に対しては感受性を示した.
おわりに
筆者らは,mTORのキナーゼドメインとFRBドメインの両方を標的として同時に結合することができる新世代のmTOR阻害薬RapaLinkを創出した.筆者らが提唱した1分子のmTORに対しRapaLink-1が2座配位するという説は,最近,クライオ電子顕微鏡により解明されたmTOR複合体1の二量体の構造において,2分子のmTORのあいだの距離がRapaLink-1の分子長よりも有意に離れていることからも支持された2).RapaLink-1の片側の要素がmTORと結合すると,適切なリンカー長で結合したもう片方の要素が結合部位の近傍にとどまり,強い結合親和力が得られる.RapaLink-1に対する耐性株を樹立するためには,ラパマイシン誘導体あるいはmTORキナーゼ阻害薬の3カ月と比較して9カ月という長い期間を必要としたことから,RapaLink-1はがん細胞が薬剤耐性を獲得するまでの期間を延長できるかもしれない.このように,新世代のmTOR阻害薬であるRapaLink-1の創出をつうじ,がんの耐性の克服への新しいアプローチが提案された.
文 献
- Chen, J., Zheng, X. F., Brown, E. J. et al.: Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin associated protein and characterization of a critical serine residue. Proc. Natl Acad. Sci. USA, 92, 4947-4951 (1995)[PubMed]
- Aylett, C. H., Sauer, E., Imseng, S. et al.: Architecture of human mTOR complex 1. Science, 351, 48-52 (2016)[PubMed]
- Wagle, N., Grabiner, B. C., Van Allen, E. M. et al.: Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med., 371, 1426-1433 (2014)[PubMed]
- Lorenz, M. C. & Heitman, J.: TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem., 270, 27531-27537 (1995)[PubMed]
- Grabiner, B. C., Nardi, V., Birsoy, K. et al.: A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov., 4, 554-563 (2014)[PubMed]
- Cerami, E., Gao, J., Dogrusoz, U. et al.: The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401-404 (2012)[PubMed]
- Yang, H., Rudge, D. G., Koos, J. D. et al.: mTOR kinase structure, mechanism and regulation. Nature, 497, 217-223 (2013)[PubMed]
- Hsieh, A. C., Liu, Y., Edlind, M. P. et al.: The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature, 485, 55-61 (2012)[PubMed]
- Marinec, P. S., Chen, L., Barr, K. J. et al.: FK506-binding protein (FKBP) partitions a modified HIV protease inhibitor into blood cells and prolongs its lifetime in vivo. Proc. Natl Acad. Sci. USA, 106, 1336-1341 (2009)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2013年 京都薬科大学大学院にて博士号取得,同年 米国California大学San Francisco校 博士研究員を経て,2014年 武田薬品工業,2016年より同 主席研究員.
研究テーマ:ケミカルバイオロジーとメディシナルケミストリーをベースにした創薬.
抱負:すぐれた医薬品の創出に挑戦し,創薬のプロフェッショナルとして成長して,医療の未来に貢献したい.
Kevan M. Shokat
米国California大学San Francisco校 教授.
研究室URL:http://shokatlab.ucsf.edu/
© 2016 岡庭正格・Kevan M. Shokat Licensed under CC 表示 2.1 日本
