SAMD9遺伝子の変異は新規の疾患であるMIRAGE症候群をひき起こし第7染色体の欠失に関与する
鳴海覚志・天野直子・石井智弘・長谷川奉延
(慶應義塾大学医学部 小児科学教室)
email:鳴海覚志,天野直子,石井智弘,長谷川奉延
DOI: 10.7875/first.author.2016.054
SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7.
Satoshi Narumi, Naoko Amano, Tomohiro Ishii, Noriyuki Katsumata, Koji Muroya, Masanori Adachi, Katsuaki Toyoshima, Yukichi Tanaka, Ryuji Fukuzawa, Kenichi Miyako, Saori Kinjo, Shouichi Ohga, Kenji Ihara, Hirosuke Inoue, Tadamune Kinjo, Toshiro Hara, Miyuki Kohno, Shiro Yamada, Hironaka Urano, Yosuke Kitagawa, Koji Tsugawa, Asumi Higa, Masakazu Miyawaki, Takahiro Okutani, Zenro Kizaki, Hiroyuki Hamada, Minako Kihara, Kentaro Shiga, Tetsuya Yamaguchi, Manabu Kenmochi, Hiroyuki Kitajima, Maki Fukami, Atsushi Shimizu, Jun Kudoh, Shinsuke Shibata, Hideyuki Okano, Noriko Miyake, Naomichi Matsumoto, Tomonobu Hasegawa
Nature Genetics, 48, 792-797 (2016)
先天性副腎皮質機能低下症は生体における恒常性の維持に必須な副腎ステロイドホルモンの欠乏する致死性の疾患であり,その成因については未解明な点が多かった.筆者らは,原因の不明な先天性副腎皮質機能低下症の小児の患者24名に対しエキソーム解析を中心に遺伝子を解析し,11名が第7染色体に位置するSAMD9遺伝子に変異をもつことを明らかにした.この変異をもつ患者は副腎の低形成のほかにも共通した造血異常,易感染性,成長障害,性腺症状,消化器症状を示し,2名は第7染色体の欠失をともなう骨髄異形成症候群を合併していた.このような症状の組合せをもつ先天性の異常はこれまで記載がなく,新規の症候群であることからMIRAGE症候群と命名した.HEK293細胞および患者に由来する皮膚線維芽細胞を用いた実験により,変異型のSAMD9は増殖を強く抑制する効果をもつこと,患者の細胞はエンドソーム系に構造的な異常をもつことが示された.単一の遺伝子の異常を病因とする新しい先天性症候群を発見したこの研究は,臨床遺伝学的に重要であっただけでなく,悪性の疾患に頻発する染色体異常の獲得の機構に関しても洞察をあたえた.
副腎皮質は恒常性の維持やストレス応答に必須な役割をはたす副腎ステロイドホルモンを分泌する.先天性副腎皮質機能低下症は副腎ステロイドホルモンの欠乏する致死性の疾患であり,先天性副腎低形成症はそのサブタイプのひとつである.臨床的には,副腎のほかに症状のない非症候性先天性副腎低形成症と,副腎のほかにも症状のある症候性先天性副腎低形成症に分類される.非症候性先天性副腎低形成症の成因としては,副腎の形成に重要な転写因子DAX1の異常,ステロイドホルモン産生能と細胞増殖能を制御する副腎皮質刺激ホルモンのシグナル受容機構の異常などが知られる.一方,症候性先天性副腎低形成症を含む症候群としてはトリプルA症候群,IMAGe症候群などが知られている.この研究は,先天性副腎皮質機能低下症の分子基盤を明らかにし,疾患の発症にかかわる新たな分子機構を探索することを目的とした.
筆者らは,1980年代から,先天性副腎皮質機能低下症の生化学診断および遺伝子診断につき研究しており,2歳未満で発症した原発性の副腎皮質機能低下症であり,生化学的な分析により副腎皮質機能低下症に特異的なパターンを示さない,という,生化学的な特徴および所見のない先天性副腎皮質機能低下症の小児の患者のゲノムDNA検体として41名分を蓄積していた.この41名につき,Sanger法あるいは次世代シークエンサーを用いたターゲットリシーケンス法により,先天性副腎皮質機能低下症に関連する11の遺伝子について解析した.26名においては既知の責任遺伝子に変異が同定されたが,残りの15名には変異が認められなかった.これら15名を原因の不明な先天性副腎皮質機能低下症の小児の患者と定義し,その分子病態を明らかにするため,6名においてエキソーム解析を行った.劣性遺伝モデルでは複数の患者のあいだで共通する候補遺伝子はなかったが,優性遺伝モデルでは4名が共通して変異をもつ遺伝子としてSAMD9遺伝子が抽出された.残りの9名,および,原因の不明な先天性副腎皮質機能低下症の小児の患者の別のコホート9名においてSAMD9遺伝子を解析したところ,さらに変異をもつ7名の患者を同定し,最終的に,SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は10家系11名に達した.これらの変異はいずれもヘテロ接合性であったが,両親について解析した7家系において両親には変異が認められず,変異の大部分は発端者の世代において新生したde novo変異と考えられた.
11名のSAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者の臨床像を詳細に検討した.副腎皮質機能低下症は日齢0~51日において発症しており,いずれも重症であった.7名において副腎の形態が評価されたが,いずれも低形成であった.剖検をうけた2名の副腎皮質は重度の低形成であった.
SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は,いずれも副腎のほかに特徴的な症状があり,症候性先天性副腎低形成症に分類された.すべての患者に出生よりまえからの発育の遅延があり,早産かつ不当軽量児として出生していた.長期に生存した例においては出生ののちにも成長の障害が持続し,いちじるしい低身長症であった.男児のすべての例において外性器に異常があった.46,XY核型ながら外性器が完全女性型であり戸籍上の性別を女性とした例が1名あった.剖検をうけた女児2名においては卵巣の重度の低形成および原始卵胞の減少がみられた.すべての患者において,乳児期の早期に輸血を要する血小板減少症あるいは貧血が認められた.これらは生後6カ月までに自然に寛解したが,2名は幼児期に造血の異常が再燃し,のちに,その原因は第7染色体の欠失をともなう骨髄異形成症候群であることがわかった.易感染性もすべての例に共通する特徴であり,侵襲性細菌感染症や日和見感染症がみられた.7名は感染症のため2歳未満で死亡した.長期に生存した例において易感染性は経年的に改善された.また,慢性下痢も共通してみられ,難治性の肛門周囲膿瘍を合併した例もあった.
SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は,副腎のほかに共通の症状をもつ症候性先天性副腎低形成症と考えられた.これまで述べたような症状の組合せをもつ先天性の異常はこれまで記載がなく,新規の疾患単位と考えられた.6つの主要な徴候,造血異常(myelodysplasia),易感染性(infection),成長障害(restriction of growth),先天性副腎低形成症(adrenal hypoplasia),性腺症状(genital phenotypes),消化器症状(enteropathy)にもとづき,この新しい疾患をMIRAGE症候群と命名した.
MIRAGE症候群の患者における中心的な症状は全身性の成長障害および副腎や卵巣などの低形成であり,いずれも細胞の増殖における不良を示唆した.このため,HEK293細胞を用いてSAMD9の発現が細胞の増殖におよぼす影響について検証した.野生型のSAMD9の発現は増殖を軽度に抑制した一方,8種類の変異型のSAMD9の発現はいずれも増殖をきわめて強く抑制した.これらの観察から,野生型のSAMD9は増殖を抑制性に制御すること,MIRAGE症候群の患者において観察されたSAMD9遺伝子の変異は本来の増殖の抑制能を増強させる機能亢進型の変異であることが明らかにされた.これらの結果は,SAMD9遺伝子は侵襲性線維腫症において発現の低下する腫瘍抑制遺伝子として同定されたこと1),SAMD9遺伝子の機能喪失型の変異はすでにほかの疾患の病因として報告されていたこと2),と矛盾しなかった.
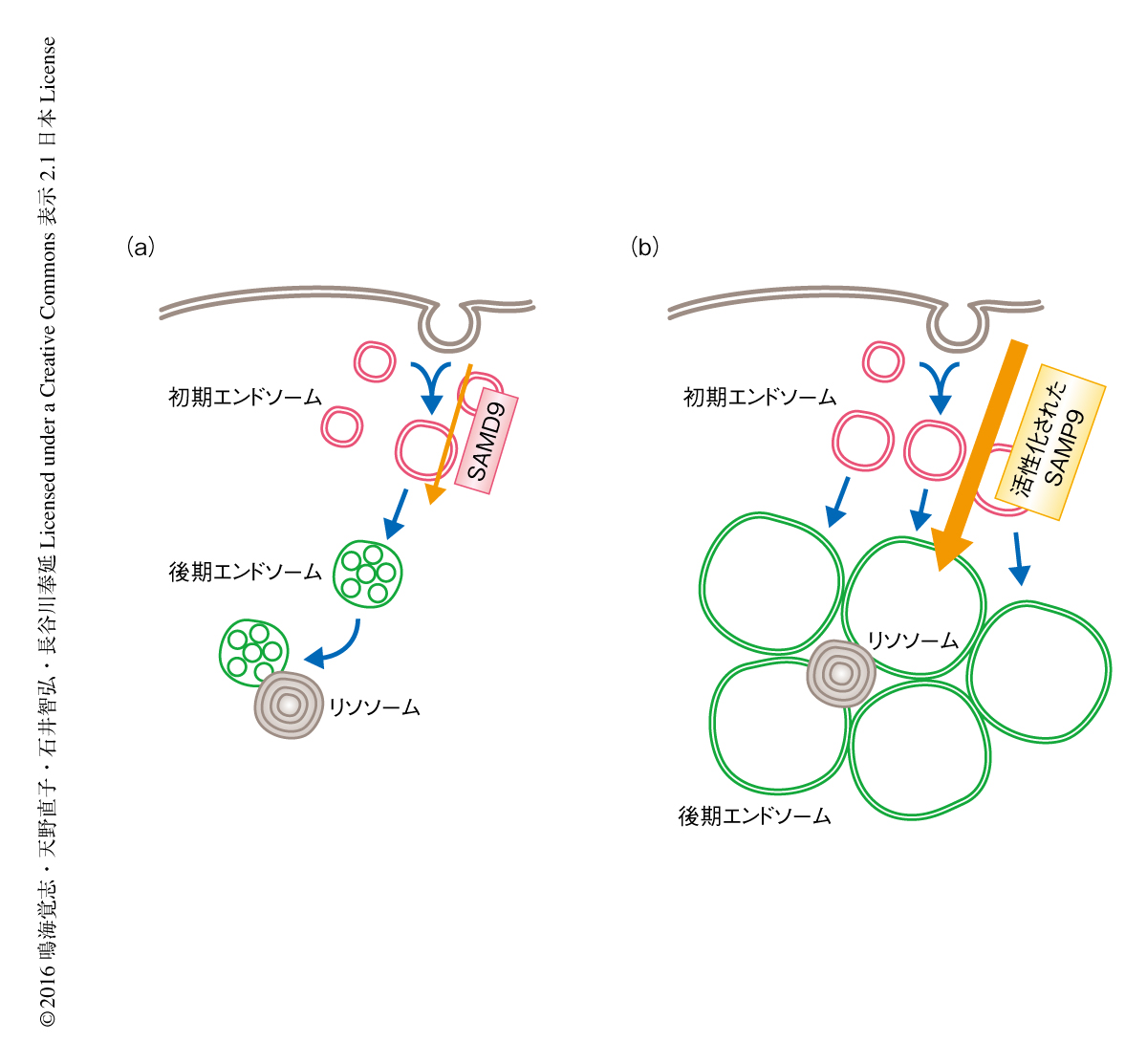
病態を細胞のレベルにおいてより詳細に解析するため,MIRAGE症候群の患者に由来する皮膚線維芽細胞を用いた.SAMD9およびそのパラログであるSAMD9Lは初期エンドソームの融合に関与するとの報告があり3),エンドソーム系に焦点をあてた.初期エンドソームをマーカーを用いて可視化したところ,患者に由来する細胞において初期エンドソームの大型化が確認された.後期エンドソームを可視化したところ,患者に由来する細胞において後期エンドソームのマーカーを発現する巨大なベシクルが細胞質を占拠するようすが観察された.しかしながら,電子顕微鏡による観察において,この巨大なベシクルは内部の構造がとぼしく,多胞体と表現される後期エンドソームに特有の顆粒の構造は認められなかった.
以上の検討から,MIRAGE症候群の患者に由来する皮膚線維芽細胞においては,初期エンドソームから後期エンドソームにいたるエンドソーム系のヒエラルキーが乱れており,初期エンドソームの大型化および後期エンドソームのマーカーをもつものの後期エンドソームとしての特徴的な内部の構造を欠いた巨大なベシクルの貯留により特徴づけられることが明らかにされた(図1).想定されるSAMD9の機能が初期エンドソームの融合であることを考慮すると,これらのエンドソーム系の変化はSAMD9の機能の亢進によりもたらされたとして矛盾のないものであった.
骨髄異形成症候群においてはさまざまな染色体に構造異常が認められるが,そのなかでも,第5染色体の長腕の欠失および第7染色体の欠失がとくに高頻度である4).MIRAGE症候群においては,11名のうち2名に第7染色体の欠失をともなう骨髄異形成症候群が合併していた.SAMD9およびそのパラログであるSAMD9Lは第7染色体に隣接してコードされている.第7染色体の欠失をともなう血液疾患である急性骨髄性白血病,骨髄異形成症候群,若年性骨髄単球性白血病における欠失領域の解析において,これらの疾患に共通して欠失する領域にはSAMD9遺伝子およびSAMD9L遺伝子が含まれており,これらの血液疾患を抑制する腫瘍抑制遺伝子であると想定されていた5).
SAMD9遺伝子に変異をもつMIRAGE症候群の患者の骨髄細胞において第7染色体が欠失した場合,変異をもつSAMD9遺伝子の存在する染色体が欠失した,あるいは,野生型のSAMD9遺伝子の存在する染色体が欠失した,の2つの可能性があった.骨髄異形成症候群に罹患した2名の骨髄塗抹標本からDNAを抽出してSAMD9遺伝子を解析したところ,変異をもつSAMD9遺伝子に由来するシグナルが大幅に低下しており,変異をもつSAMD9遺伝子の存在する染色体が欠失した可能性が支持された.また,骨髄異形成症候群を発症するまえに採取された末梢血の白血球に由来するDNAを解析したところ,臨床的な骨髄異形成症候群を発症する以前にすでに5~10%の細胞において第7染色体が欠失していた.
これらの観察を統合して,以下のストーリーが考えられた(図2).MIRAGE症候群の患者の骨髄細胞のひとつにおいて,SAMD9遺伝子に変異をもつ第7染色体が欠失した.この欠失は細胞の増殖を抑制する変異型のSAMD9を除去する効果があり,結果として,増殖において有利になった.時間の経過とともに第7染色体を欠失した細胞の割合は上昇し,最終的には,骨髄細胞の大部分をしめるにいたった.しかし,第7染色体にはSAMD9遺伝子およびSAMD9L遺伝子を含む骨髄異形成症候群の発症を抑制する複数の遺伝子が存在すると考えられており,第7染色体の欠失が骨髄細胞において骨髄異形成症候群を惹起した.

以上をより一般的な状況へと拡張すると,細胞の集団において遺伝的な異常だけでなく感染症,薬剤の使用,酸化ストレスなどを含むなんらかの理由により,細胞の増殖を抑制性に制御する遺伝子の活性が高まると,ゲノムあるいはエピゲノムの異常などによりこの遺伝子の不活性化した細胞がその特異な環境において適応的となり,クローン増殖性の疾患のリスクが増加する,というモデルを構築することが可能である.たとえば,SAMD9遺伝子の発現はインターフェロンにより誘導されることが知られており,インターフェロンの投与ののちには第7染色体の欠失に関連した血液疾患が発症しやすくなると予想することができる.この予想に合致する研究として,慢性骨髄性白血病において,インターフェロンαによる治療をうけた患者はブスルファンによる治療をうけた患者より急性転化期に第7染色体の欠失がみられるリスクが15倍も高かったという報告がある6).この現象の説明として,インターフェロンαによりSAMD9遺伝子の発現が亢進し細胞の増殖の抑制が高まった状況において,骨髄細胞において第7染色体を欠失し増殖の抑制が部分的に解除された細胞の割合が上昇し急性転化にいたった可能性がある.
さまざまな悪性の疾患においてさまざまな染色体異常が観察されるが,その獲得の機構については十分に解明されていない.SAMD9遺伝子の機能亢進型の変異をもつMIRAGE症候群において第7染色体の欠失が高頻度に観察されたことは,細胞の適応する条件の変化をつうじ染色体の欠失のリスクが高まるという特定の状況をひき起こす分子機構の実在を示すものであった.今後,個々の悪性の疾患において染色体異常の獲得が適応的となるような状況を分析することにより,まったく新しい視点からの発症あるいは進展の予防法が開発されることを期待したい.
略歴:2009年 慶應義塾大学大学院医学研究科 所定単位取得退学,2016年より国立成育医療研究センター 室長.
研究テーマ:小児における内分泌疾患の分子遺伝学的な研究.
抱負:臨床の現場に還元の可能な医学の研究を実践したい.
天野 直子(Naoko Amano)
慶應義塾大学医学部 共同研究員.
石井 智弘(Tomohiro Ishii)
慶應義塾大学医学部 専任講師.
長谷川 奉延(Tomonobu Hasegawa)
慶應義塾大学医学部 教授.
研究室URL:http://pedia.med.keio.ac.jp/
© 2016 鳴海覚志・天野直子・石井智弘・長谷川奉延 Licensed under CC 表示 2.1 日本
(慶應義塾大学医学部 小児科学教室)
email:鳴海覚志,天野直子,石井智弘,長谷川奉延
DOI: 10.7875/first.author.2016.054
SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7.
Satoshi Narumi, Naoko Amano, Tomohiro Ishii, Noriyuki Katsumata, Koji Muroya, Masanori Adachi, Katsuaki Toyoshima, Yukichi Tanaka, Ryuji Fukuzawa, Kenichi Miyako, Saori Kinjo, Shouichi Ohga, Kenji Ihara, Hirosuke Inoue, Tadamune Kinjo, Toshiro Hara, Miyuki Kohno, Shiro Yamada, Hironaka Urano, Yosuke Kitagawa, Koji Tsugawa, Asumi Higa, Masakazu Miyawaki, Takahiro Okutani, Zenro Kizaki, Hiroyuki Hamada, Minako Kihara, Kentaro Shiga, Tetsuya Yamaguchi, Manabu Kenmochi, Hiroyuki Kitajima, Maki Fukami, Atsushi Shimizu, Jun Kudoh, Shinsuke Shibata, Hideyuki Okano, Noriko Miyake, Naomichi Matsumoto, Tomonobu Hasegawa
Nature Genetics, 48, 792-797 (2016)
要 約
先天性副腎皮質機能低下症は生体における恒常性の維持に必須な副腎ステロイドホルモンの欠乏する致死性の疾患であり,その成因については未解明な点が多かった.筆者らは,原因の不明な先天性副腎皮質機能低下症の小児の患者24名に対しエキソーム解析を中心に遺伝子を解析し,11名が第7染色体に位置するSAMD9遺伝子に変異をもつことを明らかにした.この変異をもつ患者は副腎の低形成のほかにも共通した造血異常,易感染性,成長障害,性腺症状,消化器症状を示し,2名は第7染色体の欠失をともなう骨髄異形成症候群を合併していた.このような症状の組合せをもつ先天性の異常はこれまで記載がなく,新規の症候群であることからMIRAGE症候群と命名した.HEK293細胞および患者に由来する皮膚線維芽細胞を用いた実験により,変異型のSAMD9は増殖を強く抑制する効果をもつこと,患者の細胞はエンドソーム系に構造的な異常をもつことが示された.単一の遺伝子の異常を病因とする新しい先天性症候群を発見したこの研究は,臨床遺伝学的に重要であっただけでなく,悪性の疾患に頻発する染色体異常の獲得の機構に関しても洞察をあたえた.
はじめに
副腎皮質は恒常性の維持やストレス応答に必須な役割をはたす副腎ステロイドホルモンを分泌する.先天性副腎皮質機能低下症は副腎ステロイドホルモンの欠乏する致死性の疾患であり,先天性副腎低形成症はそのサブタイプのひとつである.臨床的には,副腎のほかに症状のない非症候性先天性副腎低形成症と,副腎のほかにも症状のある症候性先天性副腎低形成症に分類される.非症候性先天性副腎低形成症の成因としては,副腎の形成に重要な転写因子DAX1の異常,ステロイドホルモン産生能と細胞増殖能を制御する副腎皮質刺激ホルモンのシグナル受容機構の異常などが知られる.一方,症候性先天性副腎低形成症を含む症候群としてはトリプルA症候群,IMAGe症候群などが知られている.この研究は,先天性副腎皮質機能低下症の分子基盤を明らかにし,疾患の発症にかかわる新たな分子機構を探索することを目的とした.
1.原因の不明な先天性副腎皮質機能低下症の小児の患者に対する遺伝子解析
筆者らは,1980年代から,先天性副腎皮質機能低下症の生化学診断および遺伝子診断につき研究しており,2歳未満で発症した原発性の副腎皮質機能低下症であり,生化学的な分析により副腎皮質機能低下症に特異的なパターンを示さない,という,生化学的な特徴および所見のない先天性副腎皮質機能低下症の小児の患者のゲノムDNA検体として41名分を蓄積していた.この41名につき,Sanger法あるいは次世代シークエンサーを用いたターゲットリシーケンス法により,先天性副腎皮質機能低下症に関連する11の遺伝子について解析した.26名においては既知の責任遺伝子に変異が同定されたが,残りの15名には変異が認められなかった.これら15名を原因の不明な先天性副腎皮質機能低下症の小児の患者と定義し,その分子病態を明らかにするため,6名においてエキソーム解析を行った.劣性遺伝モデルでは複数の患者のあいだで共通する候補遺伝子はなかったが,優性遺伝モデルでは4名が共通して変異をもつ遺伝子としてSAMD9遺伝子が抽出された.残りの9名,および,原因の不明な先天性副腎皮質機能低下症の小児の患者の別のコホート9名においてSAMD9遺伝子を解析したところ,さらに変異をもつ7名の患者を同定し,最終的に,SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は10家系11名に達した.これらの変異はいずれもヘテロ接合性であったが,両親について解析した7家系において両親には変異が認められず,変異の大部分は発端者の世代において新生したde novo変異と考えられた.
2.SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者の臨床像
11名のSAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者の臨床像を詳細に検討した.副腎皮質機能低下症は日齢0~51日において発症しており,いずれも重症であった.7名において副腎の形態が評価されたが,いずれも低形成であった.剖検をうけた2名の副腎皮質は重度の低形成であった.
SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は,いずれも副腎のほかに特徴的な症状があり,症候性先天性副腎低形成症に分類された.すべての患者に出生よりまえからの発育の遅延があり,早産かつ不当軽量児として出生していた.長期に生存した例においては出生ののちにも成長の障害が持続し,いちじるしい低身長症であった.男児のすべての例において外性器に異常があった.46,XY核型ながら外性器が完全女性型であり戸籍上の性別を女性とした例が1名あった.剖検をうけた女児2名においては卵巣の重度の低形成および原始卵胞の減少がみられた.すべての患者において,乳児期の早期に輸血を要する血小板減少症あるいは貧血が認められた.これらは生後6カ月までに自然に寛解したが,2名は幼児期に造血の異常が再燃し,のちに,その原因は第7染色体の欠失をともなう骨髄異形成症候群であることがわかった.易感染性もすべての例に共通する特徴であり,侵襲性細菌感染症や日和見感染症がみられた.7名は感染症のため2歳未満で死亡した.長期に生存した例において易感染性は経年的に改善された.また,慢性下痢も共通してみられ,難治性の肛門周囲膿瘍を合併した例もあった.
SAMD9遺伝子に変異をもつ先天性副腎皮質機能低下症の患者は,副腎のほかに共通の症状をもつ症候性先天性副腎低形成症と考えられた.これまで述べたような症状の組合せをもつ先天性の異常はこれまで記載がなく,新規の疾患単位と考えられた.6つの主要な徴候,造血異常(myelodysplasia),易感染性(infection),成長障害(restriction of growth),先天性副腎低形成症(adrenal hypoplasia),性腺症状(genital phenotypes),消化器症状(enteropathy)にもとづき,この新しい疾患をMIRAGE症候群と命名した.
3.SAMD9遺伝子の変異が細胞におよぼす影響
MIRAGE症候群の患者における中心的な症状は全身性の成長障害および副腎や卵巣などの低形成であり,いずれも細胞の増殖における不良を示唆した.このため,HEK293細胞を用いてSAMD9の発現が細胞の増殖におよぼす影響について検証した.野生型のSAMD9の発現は増殖を軽度に抑制した一方,8種類の変異型のSAMD9の発現はいずれも増殖をきわめて強く抑制した.これらの観察から,野生型のSAMD9は増殖を抑制性に制御すること,MIRAGE症候群の患者において観察されたSAMD9遺伝子の変異は本来の増殖の抑制能を増強させる機能亢進型の変異であることが明らかにされた.これらの結果は,SAMD9遺伝子は侵襲性線維腫症において発現の低下する腫瘍抑制遺伝子として同定されたこと1),SAMD9遺伝子の機能喪失型の変異はすでにほかの疾患の病因として報告されていたこと2),と矛盾しなかった.
病態を細胞のレベルにおいてより詳細に解析するため,MIRAGE症候群の患者に由来する皮膚線維芽細胞を用いた.SAMD9およびそのパラログであるSAMD9Lは初期エンドソームの融合に関与するとの報告があり3),エンドソーム系に焦点をあてた.初期エンドソームをマーカーを用いて可視化したところ,患者に由来する細胞において初期エンドソームの大型化が確認された.後期エンドソームを可視化したところ,患者に由来する細胞において後期エンドソームのマーカーを発現する巨大なベシクルが細胞質を占拠するようすが観察された.しかしながら,電子顕微鏡による観察において,この巨大なベシクルは内部の構造がとぼしく,多胞体と表現される後期エンドソームに特有の顆粒の構造は認められなかった.
以上の検討から,MIRAGE症候群の患者に由来する皮膚線維芽細胞においては,初期エンドソームから後期エンドソームにいたるエンドソーム系のヒエラルキーが乱れており,初期エンドソームの大型化および後期エンドソームのマーカーをもつものの後期エンドソームとしての特徴的な内部の構造を欠いた巨大なベシクルの貯留により特徴づけられることが明らかにされた(図1).想定されるSAMD9の機能が初期エンドソームの融合であることを考慮すると,これらのエンドソーム系の変化はSAMD9の機能の亢進によりもたらされたとして矛盾のないものであった.
4.SAMD9遺伝子の変異と第7染色体の欠失との関連
骨髄異形成症候群においてはさまざまな染色体に構造異常が認められるが,そのなかでも,第5染色体の長腕の欠失および第7染色体の欠失がとくに高頻度である4).MIRAGE症候群においては,11名のうち2名に第7染色体の欠失をともなう骨髄異形成症候群が合併していた.SAMD9およびそのパラログであるSAMD9Lは第7染色体に隣接してコードされている.第7染色体の欠失をともなう血液疾患である急性骨髄性白血病,骨髄異形成症候群,若年性骨髄単球性白血病における欠失領域の解析において,これらの疾患に共通して欠失する領域にはSAMD9遺伝子およびSAMD9L遺伝子が含まれており,これらの血液疾患を抑制する腫瘍抑制遺伝子であると想定されていた5).
SAMD9遺伝子に変異をもつMIRAGE症候群の患者の骨髄細胞において第7染色体が欠失した場合,変異をもつSAMD9遺伝子の存在する染色体が欠失した,あるいは,野生型のSAMD9遺伝子の存在する染色体が欠失した,の2つの可能性があった.骨髄異形成症候群に罹患した2名の骨髄塗抹標本からDNAを抽出してSAMD9遺伝子を解析したところ,変異をもつSAMD9遺伝子に由来するシグナルが大幅に低下しており,変異をもつSAMD9遺伝子の存在する染色体が欠失した可能性が支持された.また,骨髄異形成症候群を発症するまえに採取された末梢血の白血球に由来するDNAを解析したところ,臨床的な骨髄異形成症候群を発症する以前にすでに5~10%の細胞において第7染色体が欠失していた.
これらの観察を統合して,以下のストーリーが考えられた(図2).MIRAGE症候群の患者の骨髄細胞のひとつにおいて,SAMD9遺伝子に変異をもつ第7染色体が欠失した.この欠失は細胞の増殖を抑制する変異型のSAMD9を除去する効果があり,結果として,増殖において有利になった.時間の経過とともに第7染色体を欠失した細胞の割合は上昇し,最終的には,骨髄細胞の大部分をしめるにいたった.しかし,第7染色体にはSAMD9遺伝子およびSAMD9L遺伝子を含む骨髄異形成症候群の発症を抑制する複数の遺伝子が存在すると考えられており,第7染色体の欠失が骨髄細胞において骨髄異形成症候群を惹起した.
以上をより一般的な状況へと拡張すると,細胞の集団において遺伝的な異常だけでなく感染症,薬剤の使用,酸化ストレスなどを含むなんらかの理由により,細胞の増殖を抑制性に制御する遺伝子の活性が高まると,ゲノムあるいはエピゲノムの異常などによりこの遺伝子の不活性化した細胞がその特異な環境において適応的となり,クローン増殖性の疾患のリスクが増加する,というモデルを構築することが可能である.たとえば,SAMD9遺伝子の発現はインターフェロンにより誘導されることが知られており,インターフェロンの投与ののちには第7染色体の欠失に関連した血液疾患が発症しやすくなると予想することができる.この予想に合致する研究として,慢性骨髄性白血病において,インターフェロンαによる治療をうけた患者はブスルファンによる治療をうけた患者より急性転化期に第7染色体の欠失がみられるリスクが15倍も高かったという報告がある6).この現象の説明として,インターフェロンαによりSAMD9遺伝子の発現が亢進し細胞の増殖の抑制が高まった状況において,骨髄細胞において第7染色体を欠失し増殖の抑制が部分的に解除された細胞の割合が上昇し急性転化にいたった可能性がある.
おわりに
さまざまな悪性の疾患においてさまざまな染色体異常が観察されるが,その獲得の機構については十分に解明されていない.SAMD9遺伝子の機能亢進型の変異をもつMIRAGE症候群において第7染色体の欠失が高頻度に観察されたことは,細胞の適応する条件の変化をつうじ染色体の欠失のリスクが高まるという特定の状況をひき起こす分子機構の実在を示すものであった.今後,個々の悪性の疾患において染色体異常の獲得が適応的となるような状況を分析することにより,まったく新しい視点からの発症あるいは進展の予防法が開発されることを期待したい.
文 献
- Li, C. F., MacDonald, J. R., Wei, R. Y. et al.: Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC Genomics, 8, 92 (2007)[PubMed]
- Topaz, O., Indelman, M., Chefetz, I. et al.: A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am. J. Hum. Genet., 79, 759-764 (2006)[PubMed]
- Nagamachi, A., Matsui, H., Asou, H. et al.: Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell, 24, 305-317 (2013)[PubMed]
- Haase, D., Germing, U., Schanz, J. et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood, 110, 4385-4395 (2007)[PubMed]
- Asou, H., Matsui, H., Ozaki, Y. et al.: Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochem. Biophys. Res. Commun., 383, 245-251 (2009)[PubMed]
- Tanaka, H., Tanaka, K., Oguma, N. et al.: Effect of interferon-α on chromosome abnormalities in treated chronic myelogenous leukemia patients. Cancer Genet. Cytogenet., 153, 133-143 (2004)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2009年 慶應義塾大学大学院医学研究科 所定単位取得退学,2016年より国立成育医療研究センター 室長.
研究テーマ:小児における内分泌疾患の分子遺伝学的な研究.
抱負:臨床の現場に還元の可能な医学の研究を実践したい.
天野 直子(Naoko Amano)
慶應義塾大学医学部 共同研究員.
石井 智弘(Tomohiro Ishii)
慶應義塾大学医学部 専任講師.
長谷川 奉延(Tomonobu Hasegawa)
慶應義塾大学医学部 教授.
研究室URL:http://pedia.med.keio.ac.jp/
© 2016 鳴海覚志・天野直子・石井智弘・長谷川奉延 Licensed under CC 表示 2.1 日本
