クライシスにおける細胞死はM期に細胞周期が停止しているときのテロメアの脱保護により仲介される
林 眞理
(米国Salk Institute for Biological Studies,Molecular and Cell Biology Laboratory)
email:林 眞理
DOI: 10.7875/first.author.2015.090
Cell death during crisis is mediated by mitotic telomere deprotection.
Makoto T. Hayashi, Anthony J. Cesare, Teresa Rivera, Jan Karlseder
Nature, 522, 492-496 (2015)
腫瘍の形成は複製老化およびクライシスという2つのステージにおいて抑制されている.細胞老化は短小化したテロメアによりひき起こされる.しかし,腫瘍抑制経路を構成するp53やRbに異常をもつ細胞は細胞老化を迂回して増殖をつづけクライシスにおちいる.クライシスにおいてはテロメアの融合が観察され,集団のほとんどの細胞は死にいたる.ここで生き残った細胞は腫瘍化する可能性があるため,クライシスにおける細胞死は腫瘍の抑制において重要であるが,その機構についてはよくわかっていなかった.この研究において,ヒトの細胞はクライシスにおちいるとM期において細胞周期の自発的な停止を起こし,結果として,M期に停止しているときおよびそののちの細胞周期において細胞死にいたることが見い出された.テロメアの融合はp53の機能の低下した若い細胞においてもM期の停止をひき起こしたことから,細胞死をひき起こす原因であることが示唆された.M期において細胞周期の停止した細胞にはテロメアの脱保護のみられることが知られていたが,テロメア保護タンパク質TRF2のノックダウンによりM期に停止しているときのテロメアの脱保護を亢進させたところ細胞死を起こす細胞の割合が上昇した.また,TRF2のノックダウンにより有糸分裂阻害剤に対するがん細胞の感受性が上昇した.筆者らは,これらの結果から,テロメアの融合がM期における細胞周期の停止を誘導し,M期に停止しているときのテロメアの脱保護を介して細胞死をひき起こすことにより,細胞の集団から前がん状態の細胞を取り除くというクライシスにおける細胞死の経路を提唱した.
染色体の末端は,DNAのくり返し配列とそこに特異的に結合するタンパク質からなるテロメアが構築する特殊な構造により保護されている1).細胞老化は複製にともない短小化したテロメアが末端保護機能の一部を失い脱保護されることによりひき起こされる.テロメアが脱保護されるとその末端はDNAの傷害として認識され,DNA傷害チェックポイントが活性化される.通常,染色体の切断された箇所はDNA修復機構によりつながれてもとの状態にもどる.しかし,細胞老化の原因となる脱保護されたテロメアにおいては,テロメアの末端のあいだのDNA修復機構は抑制されている2).これによりテロメアの末端は傷害として認識されつづけ,そのため細胞周期が不可逆的に停止することが細胞老化の分子機構であると考えられている.
ところが,傷害チェックポイント機構に異常をもつ細胞は細胞老化を迂回して増殖をつづけ,やがて,クライシスにおちいる.クライシスによりテロメアのさらなる短小化や欠失が進行し,テロメアの保護機能を完全に失った染色体のあいだで融合が起こり,集団のほとんどの細胞は死にいたる.同時に染色体の不安定性も蓄積するため,たとえば,テロメアを伸長する酵素テロメラーゼを活性化するなどしてテロメアの長さを維持できる細胞が現われると集団において生き残り,やがて腫瘍化にいたる可能性がある.よって,クライシスによる細胞死は腫瘍の抑制において重要であるが,その機構についてはよくわかっていなかった.
近年,筆者らは,M期において長期間にわたり細胞周期を人為的に停止させた細胞においてテロメアの脱保護がひき起こされるという,それまで知られていなかった現象を報告した3).この発見をうけ,M期における細胞周期の停止がクライシスにおいてなんらかの機能をもつのではないかと考えた.
ヒトの繊維芽細胞に由来するIMR-90細胞にヒトパピローマウイルスに由来するE6およびE7を発現させることによりクライシスを誘導し生細胞イメージング法により観察した.E6はがん抑制タンパク質p53を,E7はがん抑制タンパク質Rbを阻害する.その結果,細胞老化にいたりつつある細胞を含め正常な細胞ではほとんどの細胞がM期を2時間以内に終了したのに対し,クライシスにおちいった細胞ではM期に2時間以上かかる(これを,M期の停止と定義した)細胞の割合が上昇することがわかった.この現象は,E6のみの発現,および,p53のドミナントネガティブ変異体の発現によってもひき起こされたが,E7のみの発現ではみられなかったことから,p53の機能の低下によりM期の停止は誘導されることがわかった.一方,若くてテロメア長の長い細胞ではE6およびE7を発現させてもこのような表現型は観察されなかった.さらに,クライシスにおちいりつつあるE6およびE7を発現させた細胞やp53のドミナントネガティブ変異体を発現させた細胞におけるM期の停止はテロメラーゼの発現により抑制された.このことから,p53の機能を失いクライシスにおちいった細胞では,テロメアの短小化によりM期における細胞周期の停止がひき起こされると考えられた.
クライシスにおけるテロメアの短小化および欠失はテロメアの融合をひき起こすことから,テロメアの融合がM期における細胞周期の停止の原因であるとの仮説をたてた.これを検証するため,E6およびE7を発現させたクライシスにおちいるまえの若いIMR-90細胞において,CRISPR/Cas9系4) を用いてテロメア保護タンパク質であるTRF2をノックアウトした.TRF2の欠損はテロメアの融合をひき起こすことが知られていたが5),この細胞の生細胞イメージング法による観察によりM期の停止も誘導されていることが明らかにされた.この表現型はCRISPR/Cas9系の標的となった配列にアミノ酸変異を起こさないような変異を導入したTRF2の過剰発現により抑制されたことから,オフターゲット作用ではないことが確認された.さらに,テロメアの融合に必要であることが報告されているLIG4や53BP1といったタンパク質6,7) をTRF2と同時にノックアウトすると,テロメアの融合にくわえM期の停止も抑制された.これらの結果から,テロメアの融合がM期における細胞周期の停止をひき起こすと結論した.
筆者らは,さきにも述べたように,薬剤などによりM期において細胞周期を人為的に停止させた細胞においてテロメアの脱保護の誘導されることを報告していた3).そこで,クライシスによるM期における細胞周期の自発的な停止においても同様にテロメアの脱保護がみられるか検証した.M期の停止した細胞においては姉妹染色分体が早期に分離することが知られていたため,この表現型をM期の停止のマーカーとした.クライシスにおちいったM期の細胞を観察することにより,姉妹染色分体が早期に分離している細胞ではテロメアの脱保護が2倍以上も増加していることがわかった.この結果から,クライシスによるM期における細胞周期の自発的な停止においてもテロメアの脱保護がひき起こされることが示唆された.
クライシスによりM期において細胞周期の停止した細胞がどのような運命をたどるのか生細胞イメージング法により観察したところ,M期に停止する時間が長いほどM期において細胞死する割合が高くなることがわかった.そこで,クライシスによりどの程度の細胞がM期において細胞死するか調べたところ,M期において細胞死する細胞は約5割で,残りの細胞は間期において細胞死することが見い出された.ところが,これら間期に細胞死する細胞の直前のM期について生細胞イメージング法のデータから解析したところ,約半数の細胞はM期の停止を起こしていたことがわかった.すなわち,クライシスによる細胞死の8割近くはM期に停止しているときあるいはそのあとに起こっており,M期における細胞周期の停止がクライシスによる細胞死の主要な原因であることがつきとめられた.
残された大きな疑問は,M期に細胞周期が停止しているときのテロメアの脱保護は細胞死の原因なのかどうかであった.筆者らは,これまでの研究により,shRNAを用いてTRF2を部分的にノックダウンすると,M期に細胞周期が停止しているときのテロメアの脱保護が亢進することを明らかにしていた2).そこで,このことを利用し,IMR-90細胞においてTRF2を部分的にノックダウンして成長の速度を測定した.この細胞ではわずかに残されたTRF2がテロメアの保護機能の一部を保持しており,テロメアの融合はほとんど観察されない2).このことと一致して,この細胞はクライシスにおちいるまえは対照となる細胞と比較して遜色ない成長を示した.ところが,TRF2を部分的にノックダウンした細胞を長期間にわたり培養したところ,クライシスにおちいったときの細胞の増殖が極端に低下した.クライシスにおちいったM期の細胞の観察により,この細胞ではM期におけるテロメアの脱保護が非常に亢進していることが確認された.さらに,生細胞イメージング法によりこの細胞ではM期に停止したのちの細胞死の頻度が有意に上昇していることが明らかにされた.一方,TRF2を過剰に発現させることによりM期において停止している細胞においてテロメアの脱保護を抑制することも可能であり3),この条件においてはクライシスにおけるM期の自発的な停止における細胞死が抑制されることも確認された.これらの実験的な証拠から,M期に細胞周期が停止しているときのテロメアの脱保護が細胞死の一因であるとの仮説が支持された.
もし,この仮説が正しければ,TRF2を部分的にノックダウンした細胞はM期において細胞周期の停止をひき起こす薬剤に対し感受性が上昇していることが予想された.そこで,がん細胞に由来するHT1080 6TG細胞においてTRF2を部分的にノックダウンし,異なる作用によりM期の停止をひき起こすさまざまな薬剤に対する感受性について調べたところ,すべての場合において対照となる細胞と比較し感受性は上昇していた.この効果は,M期の停止をひき起こさずにDNA傷害をひき起こす薬剤にはみられなかった.このことから,TRF2の減少による薬剤感受性の上昇の効果はM期において細胞周期の停止をひき起こす薬剤に特異的であることがつきとめられた.
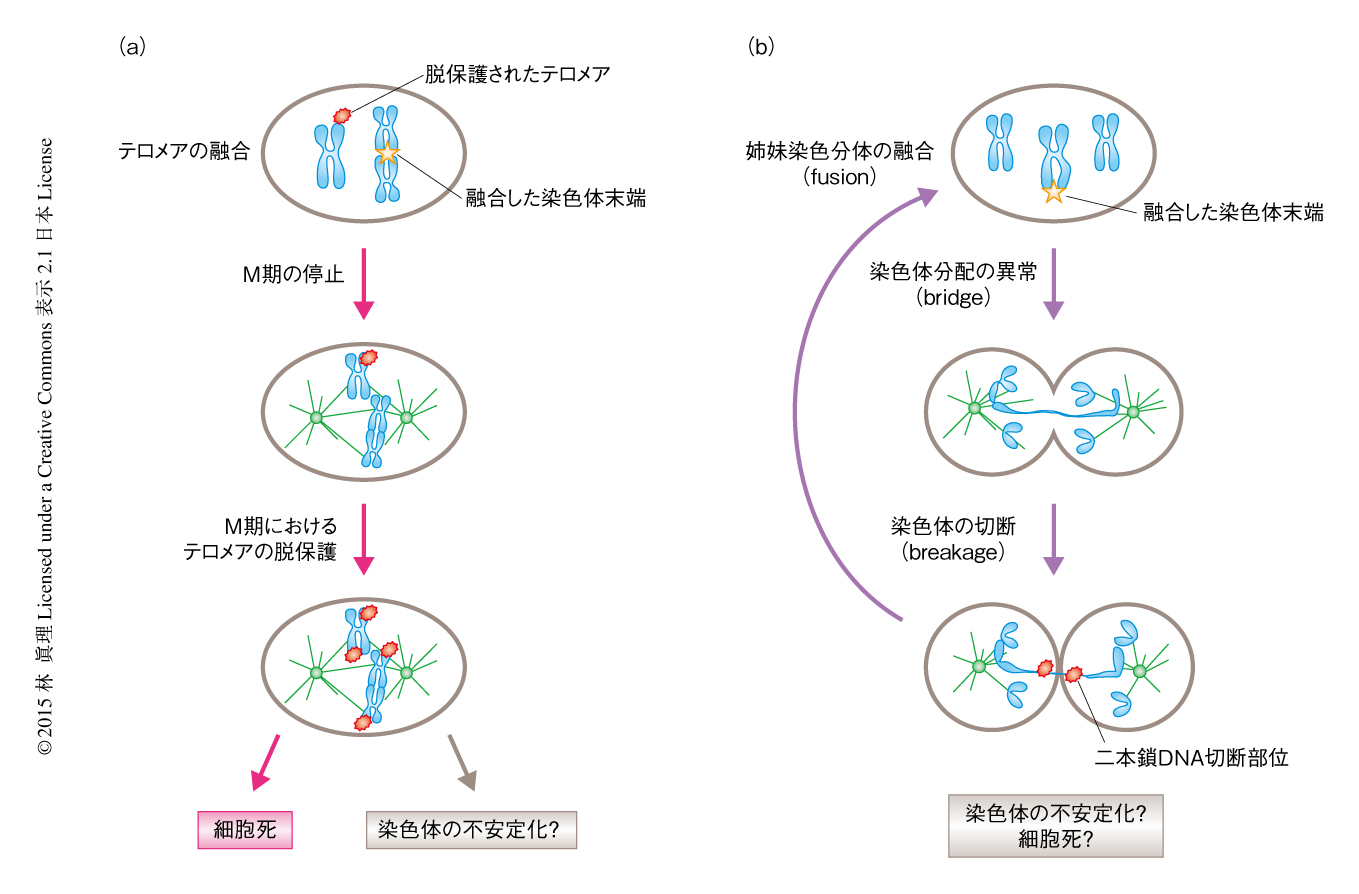
筆者らは,この研究において,クライシスにおちいった細胞において極度に短小化したテロメアが融合し,この融合によりM期における細胞周期の停止がひき起こされ,M期に停止しているときのさらなるテロメアの脱保護により細胞死が誘導される,というクライシスにおける細胞死の経路を提唱した(図1a).クライシスにおける細胞の挙動については,BFB(breakage-fusion-bridge)サイクル仮説が有名である8).放射線の照射により断裂(breakage)した姉妹染色分体どうしが融合(fusion)した場合,染色体分配のときに姉妹細胞のあいだにまたがった状態(bridge)になり,細胞質分裂のときに切断(breakage)されふたたび融合(fusion)をくり返す,というモデルである.このBFBサイクル仮説は,そののち,テロメラーゼをノックアウトしたマウスの細胞ががん化する際にテロメアの融合が観察されたことから9),ヒトのがん化における染色体の不安定化や細胞死を説明するモデルとして広く受け入れられている(図1b).筆者らの研究成果はBFBサイクル仮説を完全に否定するものではないが,少なくとも,テロメアの融合がBFBサイクルをへずにM期における細胞周期の停止を介して細胞死を誘導する経路の存在を示した.M期の停止がいくつかの抗がん剤の薬理作用であることをかんがみると,この研究がより効果的な創薬に結びつく可能性も期待される.また,M期において停止したすべての細胞が死にいたるわけではないことも明らかにされた.M期においていちど停止した細胞はのちの細胞周期において異常な小核の形成などを介して染色体の不安定性を蓄積することが報告されている10).クライシスによるM期における細胞周期の停止は細胞死のみならず染色体の不安定化をも促進することにより,がん化の抑制と促進の両方向に諸刃の剣として機能しているのかもしれない.
略歴:2008年 大阪大学大学院理学研究科にて博士号取得,2009年 米国Salk Institute for Biological Studies博士研究員を経て,2015年より京都大学白眉センター 特定助教.
研究テーマ:ヒトの体細胞におけるがん化の初期の過程.
関心事:日本の行く末.
© 2015 林 眞理 Licensed under CC 表示 2.1 日本
(米国Salk Institute for Biological Studies,Molecular and Cell Biology Laboratory)
email:林 眞理
DOI: 10.7875/first.author.2015.090
Cell death during crisis is mediated by mitotic telomere deprotection.
Makoto T. Hayashi, Anthony J. Cesare, Teresa Rivera, Jan Karlseder
Nature, 522, 492-496 (2015)
要 約
腫瘍の形成は複製老化およびクライシスという2つのステージにおいて抑制されている.細胞老化は短小化したテロメアによりひき起こされる.しかし,腫瘍抑制経路を構成するp53やRbに異常をもつ細胞は細胞老化を迂回して増殖をつづけクライシスにおちいる.クライシスにおいてはテロメアの融合が観察され,集団のほとんどの細胞は死にいたる.ここで生き残った細胞は腫瘍化する可能性があるため,クライシスにおける細胞死は腫瘍の抑制において重要であるが,その機構についてはよくわかっていなかった.この研究において,ヒトの細胞はクライシスにおちいるとM期において細胞周期の自発的な停止を起こし,結果として,M期に停止しているときおよびそののちの細胞周期において細胞死にいたることが見い出された.テロメアの融合はp53の機能の低下した若い細胞においてもM期の停止をひき起こしたことから,細胞死をひき起こす原因であることが示唆された.M期において細胞周期の停止した細胞にはテロメアの脱保護のみられることが知られていたが,テロメア保護タンパク質TRF2のノックダウンによりM期に停止しているときのテロメアの脱保護を亢進させたところ細胞死を起こす細胞の割合が上昇した.また,TRF2のノックダウンにより有糸分裂阻害剤に対するがん細胞の感受性が上昇した.筆者らは,これらの結果から,テロメアの融合がM期における細胞周期の停止を誘導し,M期に停止しているときのテロメアの脱保護を介して細胞死をひき起こすことにより,細胞の集団から前がん状態の細胞を取り除くというクライシスにおける細胞死の経路を提唱した.
はじめに
染色体の末端は,DNAのくり返し配列とそこに特異的に結合するタンパク質からなるテロメアが構築する特殊な構造により保護されている1).細胞老化は複製にともない短小化したテロメアが末端保護機能の一部を失い脱保護されることによりひき起こされる.テロメアが脱保護されるとその末端はDNAの傷害として認識され,DNA傷害チェックポイントが活性化される.通常,染色体の切断された箇所はDNA修復機構によりつながれてもとの状態にもどる.しかし,細胞老化の原因となる脱保護されたテロメアにおいては,テロメアの末端のあいだのDNA修復機構は抑制されている2).これによりテロメアの末端は傷害として認識されつづけ,そのため細胞周期が不可逆的に停止することが細胞老化の分子機構であると考えられている.
ところが,傷害チェックポイント機構に異常をもつ細胞は細胞老化を迂回して増殖をつづけ,やがて,クライシスにおちいる.クライシスによりテロメアのさらなる短小化や欠失が進行し,テロメアの保護機能を完全に失った染色体のあいだで融合が起こり,集団のほとんどの細胞は死にいたる.同時に染色体の不安定性も蓄積するため,たとえば,テロメアを伸長する酵素テロメラーゼを活性化するなどしてテロメアの長さを維持できる細胞が現われると集団において生き残り,やがて腫瘍化にいたる可能性がある.よって,クライシスによる細胞死は腫瘍の抑制において重要であるが,その機構についてはよくわかっていなかった.
近年,筆者らは,M期において長期間にわたり細胞周期を人為的に停止させた細胞においてテロメアの脱保護がひき起こされるという,それまで知られていなかった現象を報告した3).この発見をうけ,M期における細胞周期の停止がクライシスにおいてなんらかの機能をもつのではないかと考えた.
1.p53の機能の低下によりクライシスにおちいった細胞ではM期において細胞周期の自発的な停止が起こる
ヒトの繊維芽細胞に由来するIMR-90細胞にヒトパピローマウイルスに由来するE6およびE7を発現させることによりクライシスを誘導し生細胞イメージング法により観察した.E6はがん抑制タンパク質p53を,E7はがん抑制タンパク質Rbを阻害する.その結果,細胞老化にいたりつつある細胞を含め正常な細胞ではほとんどの細胞がM期を2時間以内に終了したのに対し,クライシスにおちいった細胞ではM期に2時間以上かかる(これを,M期の停止と定義した)細胞の割合が上昇することがわかった.この現象は,E6のみの発現,および,p53のドミナントネガティブ変異体の発現によってもひき起こされたが,E7のみの発現ではみられなかったことから,p53の機能の低下によりM期の停止は誘導されることがわかった.一方,若くてテロメア長の長い細胞ではE6およびE7を発現させてもこのような表現型は観察されなかった.さらに,クライシスにおちいりつつあるE6およびE7を発現させた細胞やp53のドミナントネガティブ変異体を発現させた細胞におけるM期の停止はテロメラーゼの発現により抑制された.このことから,p53の機能を失いクライシスにおちいった細胞では,テロメアの短小化によりM期における細胞周期の停止がひき起こされると考えられた.
2.テロメアの融合はM期における細胞周期の停止をひき起こす
クライシスにおけるテロメアの短小化および欠失はテロメアの融合をひき起こすことから,テロメアの融合がM期における細胞周期の停止の原因であるとの仮説をたてた.これを検証するため,E6およびE7を発現させたクライシスにおちいるまえの若いIMR-90細胞において,CRISPR/Cas9系4) を用いてテロメア保護タンパク質であるTRF2をノックアウトした.TRF2の欠損はテロメアの融合をひき起こすことが知られていたが5),この細胞の生細胞イメージング法による観察によりM期の停止も誘導されていることが明らかにされた.この表現型はCRISPR/Cas9系の標的となった配列にアミノ酸変異を起こさないような変異を導入したTRF2の過剰発現により抑制されたことから,オフターゲット作用ではないことが確認された.さらに,テロメアの融合に必要であることが報告されているLIG4や53BP1といったタンパク質6,7) をTRF2と同時にノックアウトすると,テロメアの融合にくわえM期の停止も抑制された.これらの結果から,テロメアの融合がM期における細胞周期の停止をひき起こすと結論した.
3.クライシスによるM期における細胞周期の停止はテロメアの脱保護および細胞死をひき起こす
筆者らは,さきにも述べたように,薬剤などによりM期において細胞周期を人為的に停止させた細胞においてテロメアの脱保護の誘導されることを報告していた3).そこで,クライシスによるM期における細胞周期の自発的な停止においても同様にテロメアの脱保護がみられるか検証した.M期の停止した細胞においては姉妹染色分体が早期に分離することが知られていたため,この表現型をM期の停止のマーカーとした.クライシスにおちいったM期の細胞を観察することにより,姉妹染色分体が早期に分離している細胞ではテロメアの脱保護が2倍以上も増加していることがわかった.この結果から,クライシスによるM期における細胞周期の自発的な停止においてもテロメアの脱保護がひき起こされることが示唆された.
クライシスによりM期において細胞周期の停止した細胞がどのような運命をたどるのか生細胞イメージング法により観察したところ,M期に停止する時間が長いほどM期において細胞死する割合が高くなることがわかった.そこで,クライシスによりどの程度の細胞がM期において細胞死するか調べたところ,M期において細胞死する細胞は約5割で,残りの細胞は間期において細胞死することが見い出された.ところが,これら間期に細胞死する細胞の直前のM期について生細胞イメージング法のデータから解析したところ,約半数の細胞はM期の停止を起こしていたことがわかった.すなわち,クライシスによる細胞死の8割近くはM期に停止しているときあるいはそのあとに起こっており,M期における細胞周期の停止がクライシスによる細胞死の主要な原因であることがつきとめられた.
4.M期に細胞周期が停止しているときのテロメアの脱保護は細胞死をひき起こす
残された大きな疑問は,M期に細胞周期が停止しているときのテロメアの脱保護は細胞死の原因なのかどうかであった.筆者らは,これまでの研究により,shRNAを用いてTRF2を部分的にノックダウンすると,M期に細胞周期が停止しているときのテロメアの脱保護が亢進することを明らかにしていた2).そこで,このことを利用し,IMR-90細胞においてTRF2を部分的にノックダウンして成長の速度を測定した.この細胞ではわずかに残されたTRF2がテロメアの保護機能の一部を保持しており,テロメアの融合はほとんど観察されない2).このことと一致して,この細胞はクライシスにおちいるまえは対照となる細胞と比較して遜色ない成長を示した.ところが,TRF2を部分的にノックダウンした細胞を長期間にわたり培養したところ,クライシスにおちいったときの細胞の増殖が極端に低下した.クライシスにおちいったM期の細胞の観察により,この細胞ではM期におけるテロメアの脱保護が非常に亢進していることが確認された.さらに,生細胞イメージング法によりこの細胞ではM期に停止したのちの細胞死の頻度が有意に上昇していることが明らかにされた.一方,TRF2を過剰に発現させることによりM期において停止している細胞においてテロメアの脱保護を抑制することも可能であり3),この条件においてはクライシスにおけるM期の自発的な停止における細胞死が抑制されることも確認された.これらの実験的な証拠から,M期に細胞周期が停止しているときのテロメアの脱保護が細胞死の一因であるとの仮説が支持された.
もし,この仮説が正しければ,TRF2を部分的にノックダウンした細胞はM期において細胞周期の停止をひき起こす薬剤に対し感受性が上昇していることが予想された.そこで,がん細胞に由来するHT1080 6TG細胞においてTRF2を部分的にノックダウンし,異なる作用によりM期の停止をひき起こすさまざまな薬剤に対する感受性について調べたところ,すべての場合において対照となる細胞と比較し感受性は上昇していた.この効果は,M期の停止をひき起こさずにDNA傷害をひき起こす薬剤にはみられなかった.このことから,TRF2の減少による薬剤感受性の上昇の効果はM期において細胞周期の停止をひき起こす薬剤に特異的であることがつきとめられた.
おわりに
筆者らは,この研究において,クライシスにおちいった細胞において極度に短小化したテロメアが融合し,この融合によりM期における細胞周期の停止がひき起こされ,M期に停止しているときのさらなるテロメアの脱保護により細胞死が誘導される,というクライシスにおける細胞死の経路を提唱した(図1a).クライシスにおける細胞の挙動については,BFB(breakage-fusion-bridge)サイクル仮説が有名である8).放射線の照射により断裂(breakage)した姉妹染色分体どうしが融合(fusion)した場合,染色体分配のときに姉妹細胞のあいだにまたがった状態(bridge)になり,細胞質分裂のときに切断(breakage)されふたたび融合(fusion)をくり返す,というモデルである.このBFBサイクル仮説は,そののち,テロメラーゼをノックアウトしたマウスの細胞ががん化する際にテロメアの融合が観察されたことから9),ヒトのがん化における染色体の不安定化や細胞死を説明するモデルとして広く受け入れられている(図1b).筆者らの研究成果はBFBサイクル仮説を完全に否定するものではないが,少なくとも,テロメアの融合がBFBサイクルをへずにM期における細胞周期の停止を介して細胞死を誘導する経路の存在を示した.M期の停止がいくつかの抗がん剤の薬理作用であることをかんがみると,この研究がより効果的な創薬に結びつく可能性も期待される.また,M期において停止したすべての細胞が死にいたるわけではないことも明らかにされた.M期においていちど停止した細胞はのちの細胞周期において異常な小核の形成などを介して染色体の不安定性を蓄積することが報告されている10).クライシスによるM期における細胞周期の停止は細胞死のみならず染色体の不安定化をも促進することにより,がん化の抑制と促進の両方向に諸刃の剣として機能しているのかもしれない.
文 献
- Palm, W. & de Lange, T.: How shelterin protects mammalian telomeres. Annu. Rev. Genet., 42, 301-334 (2008)[PubMed]
- Cesare, A. J., Hayashi, M. T., Crabbe, L. et al : The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol. Cell, 51, 141-155 (2013)[PubMed]
- Hayashi, M. T., Cesare, A. J., Fitzpatrick, J. A. et al.: A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol., 19, 387-394 (2012)[PubMed]
- Hsu, P. D., Lander, E. S. & Zhang, F.: Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157, 1262-1278 (2014)[PubMed]
- van Steensel, B., Smogorzewska, A. & de Lange, T.: TRF2 protects human telomeres from end-to-end fusions. Cell, 92, 401-413 (1998)[PubMed]
- Smogorzewska, A., Karlseder, J., Holtgreve-Grez, H. et al.: DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol., 12, 1635-1644 (2002)[PubMed]
- Dimitrova, N., Chen, Y. -C. M., Spector, D. L. et al.: 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature, 456, 524-528 (2008)[PubMed]
- McClintock, B.: The stability of broken ends of chromosomes in Zea mays. Genetics, 26, 234-282 (1941)[PubMed]
- Artandi, S. E., Chang, S., Lee, S.- L. et al.: Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature, 406, 641-645 (2000)[PubMed]
- Zhang, C. -Z., Spektor, A., Cornils, H. et al.: Chromothripsis from DNA damage in micronuclei. Nature, 522, 179-184 (2015)[PubMed]
著者プロフィール
略歴:2008年 大阪大学大学院理学研究科にて博士号取得,2009年 米国Salk Institute for Biological Studies博士研究員を経て,2015年より京都大学白眉センター 特定助教.
研究テーマ:ヒトの体細胞におけるがん化の初期の過程.
関心事:日本の行く末.
© 2015 林 眞理 Licensed under CC 表示 2.1 日本
