Werner症候群のモデル細胞を用いて明らかにされたヒトの老化に関連するヘテロクロマチン構造の変化
鈴木啓一郎・Juan Carlos Izpisua Belmonte
(米国Salk Institute for Biological Studies,Gene Expression Laboratory)
email:鈴木啓一郎
DOI: 10.7875/first.author.2015.064
A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging.
Weiqi Zhang, Jingyi Li, Keiichiro Suzuki, Jing Qu, Ping Wang, Junzhi Zhou, Xiaomeng Liu, Ruotong Ren, Xiuling Xu, Alejandro Ocampo, Tingting Yuan, Jiping Yang, Ying Li, Liang Shi, Dee Guan, Huize Pan, Shunlei Duan, Zhichao Ding, Mo Li, Fei Yi, Ruijun Bai, Yayu Wang, Chang Chen, Fuquan Yang, Xiaoyu Li, Zimei Wang, Emi Aizawa, April Goebl, Rupa Devi Soligalla, Pradeep Reddy, Concepcion Rodriguez Esteban, Fuchou Tang, Guang-Hui Liu, Juan Carlos Izpisua Belmonte
Science, 348, 1160-1163 (2015)
Werner症候群はWRNの機能の欠損によりひき起こされる早老症である.この研究においては,ヒトのES細胞からWerner症候群のモデル細胞を作製し間葉系幹細胞へと分化させることにより,ヒストンのメチル化の欠損やヘテロクロマチン構造の変化など,早老症の細胞レベルでの疾患の機序が解明された.また,WRNはヘテロクロマチン構造に関連するSUV39H1,HP1α,LAB2βと結合してはたらくことが明らかにされた.さらに,ヒストンのメチル化活性をもつSUV39H1を不活性化した間葉系幹細胞はWerner症候群と同様の細胞老化を示した.興味深いことに,WRNの減少やヘテロクロマチン構造の変化は高齢の健常者に由来する間葉系幹細胞においても観察された.以上の結果から,WRNはヘテロクロマチン構造の維持において重要な役割をはたし,ヒトにおける老化の機構の一端を担う可能性が示唆された.
Werner症候群は加齢が通常より早く進む早老症のひとつであり,ゲノムの安定性の維持にかかわるWRNの機能が欠損することによりひき起こされる遺伝性の疾患である.Werner症候群の患者はゲノムが不安定になることにより早老の症状をひき起こすと考えられているが,そのくわしい原因は解明されていない1).ゲノムの不安定性のほか老化をひき起こす原因のひとつとして,さまざまなモデル生物においてエピゲノムの変化が示されている2-4).ヒトにおいても,早老症の患者においてヘテロクロマチンのマーカーが消失していることが明らかにされた5-7).しかしながら,Werner症候群とエピゲノムの関係については明らかにされていない.
この研究においては,ゲノムを自由に改変できるゲノム編集技術を用いて,ヒトのES細胞においてWRN遺伝子の両方の対立遺伝子を破壊することによりWerner症候群のモデル細胞を作製した.培養皿において継代をくり返すことで老化を模倣することにより,これまで考えられていたゲノムの不安定性とは別に,エピゲノムの変化が細胞老化をひき起こすことが示唆された.また,このエピゲノムの変化はWerner症候群のみならず,健常者の老化にも関連することが明らかにされた.
iPS細胞はES細胞と同様にあらゆる細胞に分化できる多能性を保ちつつ無限に増殖させることのできる多能性幹細胞である.このため,患者の体細胞からiPS細胞を作製することにより患者に由来するさまざまな細胞を大量に分化させることができるようになり,疾患の機序の解明や創薬スクリーニングへの応用が期待されている.Werner症候群の患者の体細胞からiPS細胞を作製することによりモデル細胞を作製することも可能であったが,Werner症候群の原因遺伝子にコードされるWRNはゲノムの安定性の維持に重要なはたらきをもつことからWerner症候群の患者に由来する体細胞はすでに染色体異常やほかの変異をもつことが多く,そこから作製したiPS細胞はモデル細胞としてはあまり適さなかった.そこで,ヒトの野生型のES細胞を用い,WRN遺伝子の両方の対立遺伝子を相同組換えにより破壊することによりWRNの機能を欠損させたWerner症候群のモデル細胞を作製した(図1).ゲノム編集技術によるES細胞の相同組換えには安全なアデノウイルスベクターによる高効率な遺伝子ターゲッティング法を用いた8).野生型のES細胞とそこから作製したWerner症候群のモデル細胞は遺伝的な背景が同一であるため厳密な対照になる.これらの2つの細胞株を比較することにより,WRN遺伝子の欠損によるWerner症候群の表現型を培養皿において再現した.
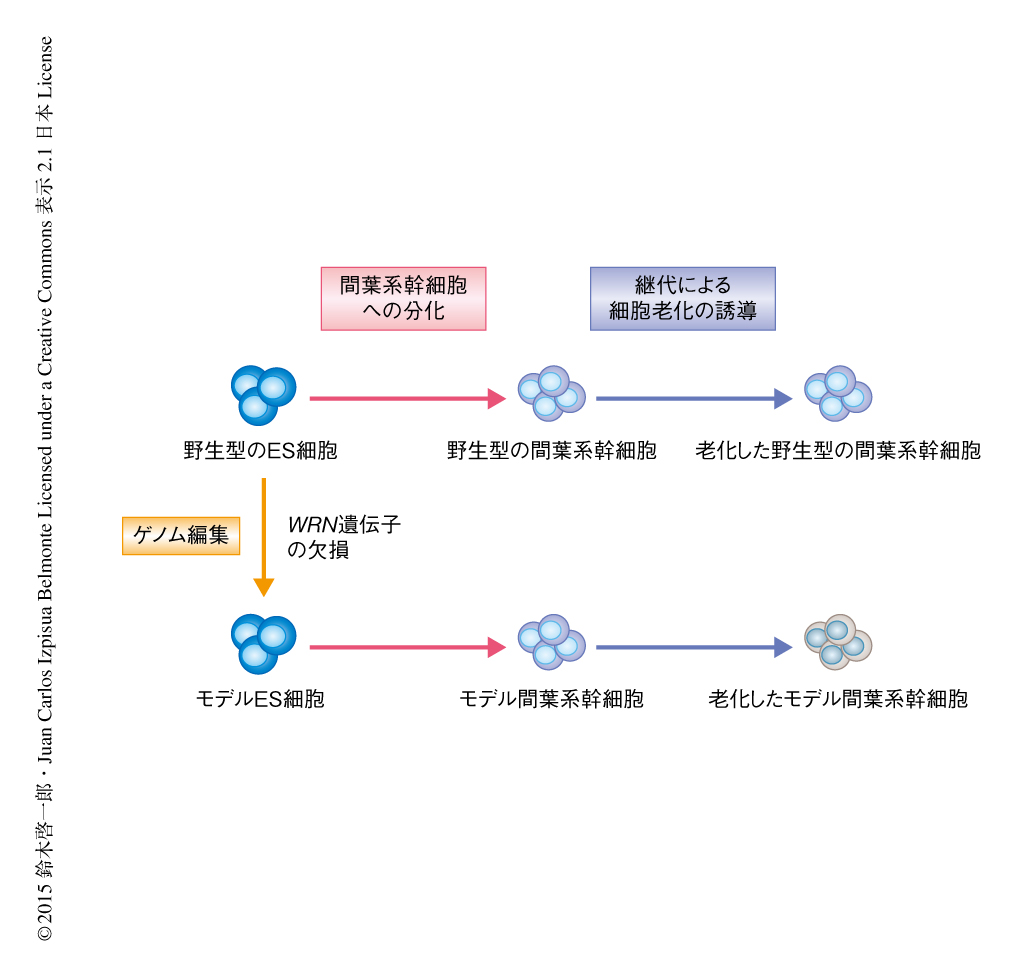
Werner症候群の患者は骨粗鬆症やアテローム性動脈硬化症など,おもに中胚葉に由来する組織において加齢にともなう症状を示す1).Werner症候群の患者は加齢にともない間葉系幹細胞が枯渇することによりさまざまな表現型を示すと考え,Werner症候群のモデルES細胞から間葉系幹細胞を分化させた.モデルES細胞は継代をくり返しても細胞老化をひき起こさず明らかな表現型は示さなかったが,そこから分化させたモデル間葉系幹細胞は継代とともに増殖能が低下し,細胞老化の指標であるβガラクトシダーゼ陽性細胞の増加,p16INK4およびp21Waf1の高発現がみられた.また,モデル間葉系幹細胞を免疫不全マウスの筋組織に移植したところ,野生型のES細胞から分化させた間葉系幹細胞に比べ,生着したのちの増殖能が低下していた.以上の結果から,間葉系幹細胞においてはWRNの欠損により細胞老化が促進されることが明らかにされた.
エピゲノムの変化は老化のひとつの指標として知られている2).そこで,Werner症候群のモデル間葉系幹細胞においてエピゲノムの変化を調べたところ,継代とともにヘテロクロマチンを核膜に局在させるLAP2βやLBRの発現の低下および核の膨張がみられ,さらに,ヘテロクロマチン構造が変化することが明らかにされた.モデル間葉系幹細胞において,ヘテロクロマチンのマーカーであるヒストンH3のLys9のトリメチル化は減少していた一方,DNAのCpG配列のメチル化およびユークロマチンのマーカーであるヒストンH3のLys4のトリメチル化は野生型のES細胞から分化させた間葉系幹細胞と同じ程度であった.ChIP-seq法により,野生型の間葉系幹細胞において20 kb以上もの大きな73カ所のヘテロクロマチン領域が同定されたが,モデル間葉系幹細胞ではそのうち28カ所(38%)が消失していた.興味深いことに,消失したヘテロクロマチン領域の86%はサブテロメア領域およびサブセントロメア領域にあった.RNA-seq法により,モデル間葉系幹細胞において発現の変化していた1047個の遺伝子のうちもっとも発現が低下していたのはセントロメアのパッケージングにかかわるタンパク質の遺伝子および核膜構成タンパク質の遺伝子であることがわかった.WRNはテロメア長の維持にかかわることが知られていたが9),モデル間葉系幹細胞においてテロメアを維持するテロメラーゼの活性が低下しテロメアは短くなっていた.以上の結果から,Werner症候群のモデル間葉系幹細胞では継代とともに核膜およびセントロメアを構成するタンパク質の発現が低下し,サブテロメア領域およびサブセントロメア領域を中心にヘテロクロマチン構造が消失することが明らかにされた.
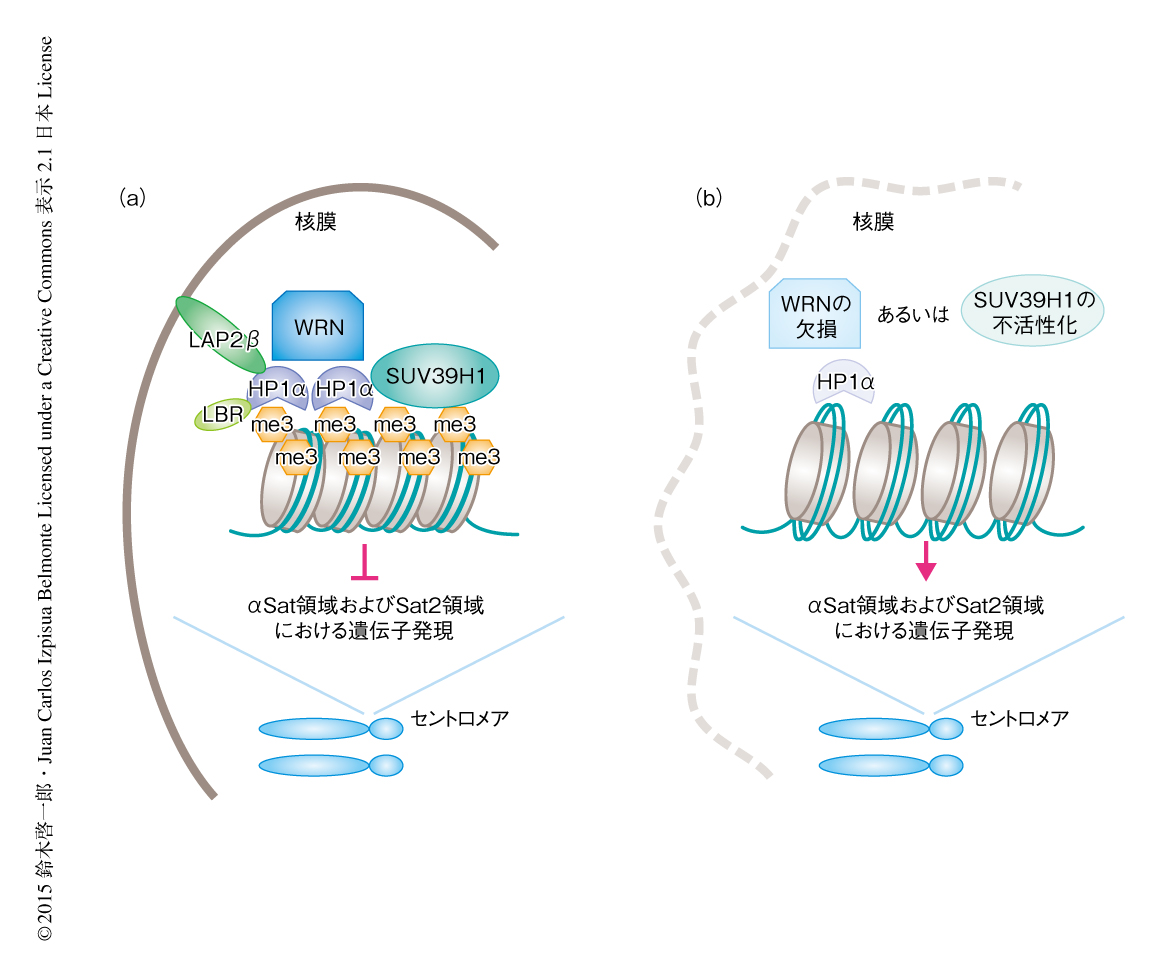
ChIP-定量PCR法により,WRNはヒストンH3のLys9のトリメチル化の集積するセントロメア領域にあるくり返し配列であるαSat領域およびSat2領域と結合することが明らかにされた.Werner症候群のモデル間葉系幹細胞ではこれらの結合がなくなりセントロメア領域においてヘテロクロマチン構造が消失することから,継代とともにαSat領域およびSat2領域からの遺伝子の発現が上昇し,また,クロマチンの構造がゆるむことにより2本鎖DNA切断が起こりやすくなりその指標であるヒストンγH2AXのシグナルがセントロメア領域において観察されるようになった.また,共免疫沈降法により,WRNは,ヒストンH3のLys9のトリメチル化活性をもつヒストンメチルトランスフェラーゼであるSUV39H1,ヘテロクロマチン領域においてヒストンH3のLys9のトリメチル化と結合するHP1α,核膜構成タンパク質でHP1αとの結合を介してヘテロクロマチン領域を核膜に局在させるLAP2βと結合することが明らかにされた.これらのことから,WRNはSUV39H1やHP1αとともにヘテロクロマチン構造の安定化にかかわることが示唆された.
ヘテロクロマチン構造の変化が細胞老化をひき起こす直接の原因を調べるため,野生型のES細胞から分化させた間葉系幹細胞においてSUV39H1あるいはHP1αをshRNAによりノックダウンした.その結果,ヒストンH3のLys9のトリメチル化は低下し,細胞老化の指標であるβガラクトシダーゼ陽性細胞の増加,p16INK4およびp21Waf1の高発現がみられ,Werner症候群のモデル間葉系幹細胞と同様の表現型を示した.逆に,モデル間葉系幹細胞においてHP1αを過剰に発現させると細胞老化は軽減された.さらに,ヒトのES細胞においてゲノム編集技術によりSUV39H1遺伝子のメチル化活性部位に点変異を導入し,間葉系幹細胞に分化させて継代をくり返したところ,モデル間葉系幹細胞と同様のヘテロクロマチン構造の変化および細胞老化を示した.ES細胞においては生殖細胞系列に特異的なヒストンメチルトランスフェラーゼであるSUV39H2の発現が高いが,間葉系幹細胞においてSUV39H2は発現していない.このため,SUV39H1遺伝子に変異を導入したES細胞においてはSUV39H2がこれを相補するためヘテロクロマチン構造に変化はみられなかったが,SUV39H2が発現しない間葉系幹細胞においてはヘテロクロマチン構造の変化とそれにともなう細胞老化がみられたと考えられた.
Werner症候群のモデル細胞の培養皿におけるヘテロクロマチン構造の変化は,健常者の老化にともないみられる一般的な現象であるかどうかを調べるため,さまざまな年齢の健常者から間葉系幹細胞を作製した.その結果,年齢の高い健常者に由来する間葉系幹細胞ではWRNの発現が低く,モデル間葉系幹細胞においてみられたような,ヒストンH3のLys9のトリメチル化の低下,および,SUV39H1,HP1α,LAP2βの発現の低下が観察され,ヘテロクロマチン構造が変化していることが示唆された.つまり,間葉系幹細胞におけるヘテロクロマチン構造の変化はWerner症候群および健常者の老化の両方にみられる共通の現象であることが明らかにされた(図2).
この研究において,ヒトのES細胞にゲノム編集技術を適応することによりWerner症候群のモデル細胞を作製し,Werner症候群の患者のみならず健常者においても,WRNのヘテロクロマチン構造の維持をとおした老化に対する役割が明らかにされた.今回は間葉系幹細胞において老化現象の一端が明らかにされたが,これを新たな足がかりとして,今後,複雑なヒトの老化現象の全体像や老化に依存的な疾患の機序の解明が期待される.
略歴:2005年 埼玉大学大学院理工学研究科 修了,2006年 埼玉医科大学 特任研究員,2009年 同 助教を経て,2010年より米国Salk Institute for Biological StudiesにてResearch Associate.
研究テーマ:ヒトの遺伝性疾患のゲノム編集技術を用いた分子機構の解明,および,新規のゲノム編集技術の開発.
Juan Carlos Izpisua Belmonte
米国Salk Institute for Biological StudiesにてProfessor.
研究室URL:http://www.salk.edu/labs/belmonte/
© 2015 鈴木啓一郎・Juan Carlos Izpisua Belmonte Licensed under CC 表示 2.1 日本
(米国Salk Institute for Biological Studies,Gene Expression Laboratory)
email:鈴木啓一郎
DOI: 10.7875/first.author.2015.064
A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging.
Weiqi Zhang, Jingyi Li, Keiichiro Suzuki, Jing Qu, Ping Wang, Junzhi Zhou, Xiaomeng Liu, Ruotong Ren, Xiuling Xu, Alejandro Ocampo, Tingting Yuan, Jiping Yang, Ying Li, Liang Shi, Dee Guan, Huize Pan, Shunlei Duan, Zhichao Ding, Mo Li, Fei Yi, Ruijun Bai, Yayu Wang, Chang Chen, Fuquan Yang, Xiaoyu Li, Zimei Wang, Emi Aizawa, April Goebl, Rupa Devi Soligalla, Pradeep Reddy, Concepcion Rodriguez Esteban, Fuchou Tang, Guang-Hui Liu, Juan Carlos Izpisua Belmonte
Science, 348, 1160-1163 (2015)
要 約
Werner症候群はWRNの機能の欠損によりひき起こされる早老症である.この研究においては,ヒトのES細胞からWerner症候群のモデル細胞を作製し間葉系幹細胞へと分化させることにより,ヒストンのメチル化の欠損やヘテロクロマチン構造の変化など,早老症の細胞レベルでの疾患の機序が解明された.また,WRNはヘテロクロマチン構造に関連するSUV39H1,HP1α,LAB2βと結合してはたらくことが明らかにされた.さらに,ヒストンのメチル化活性をもつSUV39H1を不活性化した間葉系幹細胞はWerner症候群と同様の細胞老化を示した.興味深いことに,WRNの減少やヘテロクロマチン構造の変化は高齢の健常者に由来する間葉系幹細胞においても観察された.以上の結果から,WRNはヘテロクロマチン構造の維持において重要な役割をはたし,ヒトにおける老化の機構の一端を担う可能性が示唆された.
はじめに
Werner症候群は加齢が通常より早く進む早老症のひとつであり,ゲノムの安定性の維持にかかわるWRNの機能が欠損することによりひき起こされる遺伝性の疾患である.Werner症候群の患者はゲノムが不安定になることにより早老の症状をひき起こすと考えられているが,そのくわしい原因は解明されていない1).ゲノムの不安定性のほか老化をひき起こす原因のひとつとして,さまざまなモデル生物においてエピゲノムの変化が示されている2-4).ヒトにおいても,早老症の患者においてヘテロクロマチンのマーカーが消失していることが明らかにされた5-7).しかしながら,Werner症候群とエピゲノムの関係については明らかにされていない.
この研究においては,ゲノムを自由に改変できるゲノム編集技術を用いて,ヒトのES細胞においてWRN遺伝子の両方の対立遺伝子を破壊することによりWerner症候群のモデル細胞を作製した.培養皿において継代をくり返すことで老化を模倣することにより,これまで考えられていたゲノムの不安定性とは別に,エピゲノムの変化が細胞老化をひき起こすことが示唆された.また,このエピゲノムの変化はWerner症候群のみならず,健常者の老化にも関連することが明らかにされた.
1.Werner症候群のモデルES細胞の作製
iPS細胞はES細胞と同様にあらゆる細胞に分化できる多能性を保ちつつ無限に増殖させることのできる多能性幹細胞である.このため,患者の体細胞からiPS細胞を作製することにより患者に由来するさまざまな細胞を大量に分化させることができるようになり,疾患の機序の解明や創薬スクリーニングへの応用が期待されている.Werner症候群の患者の体細胞からiPS細胞を作製することによりモデル細胞を作製することも可能であったが,Werner症候群の原因遺伝子にコードされるWRNはゲノムの安定性の維持に重要なはたらきをもつことからWerner症候群の患者に由来する体細胞はすでに染色体異常やほかの変異をもつことが多く,そこから作製したiPS細胞はモデル細胞としてはあまり適さなかった.そこで,ヒトの野生型のES細胞を用い,WRN遺伝子の両方の対立遺伝子を相同組換えにより破壊することによりWRNの機能を欠損させたWerner症候群のモデル細胞を作製した(図1).ゲノム編集技術によるES細胞の相同組換えには安全なアデノウイルスベクターによる高効率な遺伝子ターゲッティング法を用いた8).野生型のES細胞とそこから作製したWerner症候群のモデル細胞は遺伝的な背景が同一であるため厳密な対照になる.これらの2つの細胞株を比較することにより,WRN遺伝子の欠損によるWerner症候群の表現型を培養皿において再現した.
2.Werner症候群のモデル間葉系幹細胞の作製
Werner症候群の患者は骨粗鬆症やアテローム性動脈硬化症など,おもに中胚葉に由来する組織において加齢にともなう症状を示す1).Werner症候群の患者は加齢にともない間葉系幹細胞が枯渇することによりさまざまな表現型を示すと考え,Werner症候群のモデルES細胞から間葉系幹細胞を分化させた.モデルES細胞は継代をくり返しても細胞老化をひき起こさず明らかな表現型は示さなかったが,そこから分化させたモデル間葉系幹細胞は継代とともに増殖能が低下し,細胞老化の指標であるβガラクトシダーゼ陽性細胞の増加,p16INK4およびp21Waf1の高発現がみられた.また,モデル間葉系幹細胞を免疫不全マウスの筋組織に移植したところ,野生型のES細胞から分化させた間葉系幹細胞に比べ,生着したのちの増殖能が低下していた.以上の結果から,間葉系幹細胞においてはWRNの欠損により細胞老化が促進されることが明らかにされた.
3.Werner症候群のモデル間葉系幹細胞におけるヘテロクロマチン構造の変化
エピゲノムの変化は老化のひとつの指標として知られている2).そこで,Werner症候群のモデル間葉系幹細胞においてエピゲノムの変化を調べたところ,継代とともにヘテロクロマチンを核膜に局在させるLAP2βやLBRの発現の低下および核の膨張がみられ,さらに,ヘテロクロマチン構造が変化することが明らかにされた.モデル間葉系幹細胞において,ヘテロクロマチンのマーカーであるヒストンH3のLys9のトリメチル化は減少していた一方,DNAのCpG配列のメチル化およびユークロマチンのマーカーであるヒストンH3のLys4のトリメチル化は野生型のES細胞から分化させた間葉系幹細胞と同じ程度であった.ChIP-seq法により,野生型の間葉系幹細胞において20 kb以上もの大きな73カ所のヘテロクロマチン領域が同定されたが,モデル間葉系幹細胞ではそのうち28カ所(38%)が消失していた.興味深いことに,消失したヘテロクロマチン領域の86%はサブテロメア領域およびサブセントロメア領域にあった.RNA-seq法により,モデル間葉系幹細胞において発現の変化していた1047個の遺伝子のうちもっとも発現が低下していたのはセントロメアのパッケージングにかかわるタンパク質の遺伝子および核膜構成タンパク質の遺伝子であることがわかった.WRNはテロメア長の維持にかかわることが知られていたが9),モデル間葉系幹細胞においてテロメアを維持するテロメラーゼの活性が低下しテロメアは短くなっていた.以上の結果から,Werner症候群のモデル間葉系幹細胞では継代とともに核膜およびセントロメアを構成するタンパク質の発現が低下し,サブテロメア領域およびサブセントロメア領域を中心にヘテロクロマチン構造が消失することが明らかにされた.
4.セントロメア領域のヘテロクロマチン構造におけるWRNの役割
ChIP-定量PCR法により,WRNはヒストンH3のLys9のトリメチル化の集積するセントロメア領域にあるくり返し配列であるαSat領域およびSat2領域と結合することが明らかにされた.Werner症候群のモデル間葉系幹細胞ではこれらの結合がなくなりセントロメア領域においてヘテロクロマチン構造が消失することから,継代とともにαSat領域およびSat2領域からの遺伝子の発現が上昇し,また,クロマチンの構造がゆるむことにより2本鎖DNA切断が起こりやすくなりその指標であるヒストンγH2AXのシグナルがセントロメア領域において観察されるようになった.また,共免疫沈降法により,WRNは,ヒストンH3のLys9のトリメチル化活性をもつヒストンメチルトランスフェラーゼであるSUV39H1,ヘテロクロマチン領域においてヒストンH3のLys9のトリメチル化と結合するHP1α,核膜構成タンパク質でHP1αとの結合を介してヘテロクロマチン領域を核膜に局在させるLAP2βと結合することが明らかにされた.これらのことから,WRNはSUV39H1やHP1αとともにヘテロクロマチン構造の安定化にかかわることが示唆された.
5.ヘテロクロマチン構造の変化の細胞老化への影響
ヘテロクロマチン構造の変化が細胞老化をひき起こす直接の原因を調べるため,野生型のES細胞から分化させた間葉系幹細胞においてSUV39H1あるいはHP1αをshRNAによりノックダウンした.その結果,ヒストンH3のLys9のトリメチル化は低下し,細胞老化の指標であるβガラクトシダーゼ陽性細胞の増加,p16INK4およびp21Waf1の高発現がみられ,Werner症候群のモデル間葉系幹細胞と同様の表現型を示した.逆に,モデル間葉系幹細胞においてHP1αを過剰に発現させると細胞老化は軽減された.さらに,ヒトのES細胞においてゲノム編集技術によりSUV39H1遺伝子のメチル化活性部位に点変異を導入し,間葉系幹細胞に分化させて継代をくり返したところ,モデル間葉系幹細胞と同様のヘテロクロマチン構造の変化および細胞老化を示した.ES細胞においては生殖細胞系列に特異的なヒストンメチルトランスフェラーゼであるSUV39H2の発現が高いが,間葉系幹細胞においてSUV39H2は発現していない.このため,SUV39H1遺伝子に変異を導入したES細胞においてはSUV39H2がこれを相補するためヘテロクロマチン構造に変化はみられなかったが,SUV39H2が発現しない間葉系幹細胞においてはヘテロクロマチン構造の変化とそれにともなう細胞老化がみられたと考えられた.
6.細胞老化の分子指標としてのヘテロクロマチン構造の変化
Werner症候群のモデル細胞の培養皿におけるヘテロクロマチン構造の変化は,健常者の老化にともないみられる一般的な現象であるかどうかを調べるため,さまざまな年齢の健常者から間葉系幹細胞を作製した.その結果,年齢の高い健常者に由来する間葉系幹細胞ではWRNの発現が低く,モデル間葉系幹細胞においてみられたような,ヒストンH3のLys9のトリメチル化の低下,および,SUV39H1,HP1α,LAP2βの発現の低下が観察され,ヘテロクロマチン構造が変化していることが示唆された.つまり,間葉系幹細胞におけるヘテロクロマチン構造の変化はWerner症候群および健常者の老化の両方にみられる共通の現象であることが明らかにされた(図2).
おわりに
この研究において,ヒトのES細胞にゲノム編集技術を適応することによりWerner症候群のモデル細胞を作製し,Werner症候群の患者のみならず健常者においても,WRNのヘテロクロマチン構造の維持をとおした老化に対する役割が明らかにされた.今回は間葉系幹細胞において老化現象の一端が明らかにされたが,これを新たな足がかりとして,今後,複雑なヒトの老化現象の全体像や老化に依存的な疾患の機序の解明が期待される.
文 献
- Kudlow, B. A., Kennedy, B. K. & Monnat, R. J. Jr.: Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat. Rev. Mol. Cell Biol., 8, 394-404 (2007)[PubMed]
- Lopez-Otin, C., Blasco, M. A., Partridge, L. et al.: The hallmarks of aging. Cell, 153, 1194-1217 (2013)[PubMed]
- Pegoraro, G., Kubben, N., Wickert, U. et al.: Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol., 11, 1261-1267 (2009)[PubMed]
- Greer, E. L., Maures, T. J., Hauswirth, A. G. et al.: Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature, 466, 383-387 (2010)[PubMed]
- Liu, G. H., Barkho, B. Z., Ruiz, S. et al.: Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature, 472, 221-225 (2011)[PubMed]
- Shumaker, D. K., Dechat, T., Kohlmaier, A. et al.: Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA, 103, 8703-8708 (2006)[PubMed]
- Miller, J. D., Ganat, Y. M., Kishinevsky, S. et al.: Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell, 13, 691-705 (2013)[PubMed]
- Suzuki, K., Mitsui, K., Aizawa, E. et al.: Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc. Natl. Acad. Sci. USA, 105, 13781-13786 (2008)[PubMed]
- Multani, A. S. & Chang, S.: WRN at telomeres: implications for aging and cancer. J. Cell Sci., 120, 713-721 (2007)[PubMed]
著者プロフィール
略歴:2005年 埼玉大学大学院理工学研究科 修了,2006年 埼玉医科大学 特任研究員,2009年 同 助教を経て,2010年より米国Salk Institute for Biological StudiesにてResearch Associate.
研究テーマ:ヒトの遺伝性疾患のゲノム編集技術を用いた分子機構の解明,および,新規のゲノム編集技術の開発.
Juan Carlos Izpisua Belmonte
米国Salk Institute for Biological StudiesにてProfessor.
研究室URL:http://www.salk.edu/labs/belmonte/
© 2015 鈴木啓一郎・Juan Carlos Izpisua Belmonte Licensed under CC 表示 2.1 日本
